Gli errori di monitoraggio ambientale nella produzione farmaceutica sono riconducibili all'esecuzione della pulizia nel 60% dei casi. Una lettera di avvertimento della FDA del 2025 indirizzata a Excelvision Fareva citava specificamente “l’incapacità di stabilire un sistema adeguato per la pulizia e la disinfezione delle stanze e delle attrezzature per produrre condizioni asettiche”, non perché la struttura mancasse di strumenti di pulizia, ma perché le SOP non riuscivano a integrare la validazione della pulizia con i requisiti della strategia di controllo della contaminazione (CCS), lasciavano la sterilizzazione dei componenti critici non specificata e non offrivano un quadro di verifica per consentire agli operatori di dimostrare le condizioni asettiche dopo la pulizia.
Quando si verificano escursioni di monitoraggio ambientale, quando la FDA emette 483 osservazioni sulla “convalida inadeguata della pulizia” o quando gli ispettori dell’allegato 1 GMP dell’UE notano “programmi di disinfezione non convalidati”, la causa principale raramente è lo spazzolone stesso. È la procedura operativa standard che non riesce a definire la logica di classificazione dell'area, omette le specifiche del tempo di contatto, non fornisce punti di controllo di convalida e lascia gli operatori incerti se una superficie sia stata veramente pulita o semplicemente strofinata.
Questa guida fornisce il quadro normativo, la struttura passo passo, i punti di controllo di convalida e i modelli pratici necessari per scrivere una SOP per la pulizia delle camere bianche che soddisfi i requisiti ISO 14644, EU GMP Annex 1 e FDA CGMP: una POS che il QA può difendere durante le ispezioni, che gli operatori possono eseguire in modo coerente e che i team di convalida possono qualificarsi con criteri di accettazione misurabili. Che tu stia scrivendo la tua prima SOP per la pulizia farmaceutica, aggiornando le procedure legacy per soddisfare i requisiti CCS dell'Allegato 1 o standardizzando i protocolli tra più strutture asettiche, questo riferimento completo fornisce la struttura e l'approfondimento tecnico richiesti per i programmi di lavaggio conformi alle GMP negli ambienti farmaceutici ISO 5–8.
Perché le SOP per la pulizia delle camere bianche sono fondamentali ai sensi della norma ISO & GMP
Le procedure di pulizia nelle camere bianche farmaceutiche operano sotto un onere normativo unico. A differenza dei protocolli di pulizia negli edifici adibiti ad uffici, dove “abbastanza pulito” è un giudizio soggettivo, le SOP per il lavaggio farmaceutico sono documenti di prova legale che devono dimostrare, attraverso la convalida e il monitoraggio di routine, che le attività di pulizia raggiungono e mantengono lo stato ambientale classificato richiesto per la produzione di farmaci sterili.
L'allegato 1 delle GMP dell'UE (in vigore da agosto 2023) designa esplicitamente la pulizia e la disinfezione come un "elemento critico" della strategia di controllo della contaminazione (sezioni 4.10, 4.22, 4.33-4.36). L'allegato rivisto richiede che i programmi di pulizia siano convalidati, che l'efficacia della disinfezione sia monitorata regolarmente e che i disinfettanti e gli strumenti di pulizia utilizzati nelle aree asettiche di grado A/B siano sterili prima dell'uso. Questa non è una guida, ma un mandato normativo che le strutture devono implementare attraverso SOP documentate e convalidate.
La norma ISO 14644-5 (revisione 2025) richiede un programma di controllo delle operazioni (OCP) che copra esplicitamente la pulizia, il movimento del personale e il monitoraggio, con procedure “adatte ai livelli di pulizia specificati”. La norma ISO 14644-1 definisce tali livelli di pulizia in base alla concentrazione di particelle (ad esempio, Classe ISO 5: 3.520 particelle/m³ a ≥ 0,5 µm) e le attività di pulizia non devono violare tali limiti durante l'esecuzione o impedire il tempestivo ritorno alla classificazione dopo la pulizia.
Le lettere di avvertimento della FDA del 2023-2025 rivelano dove le SOP falliscono nella pratica. La lettera di avvertimento di Excelvision Fareva del 2025 citava la mancata sterilizzazione delle apparecchiature prima dell’installazione nelle linee di riempimento ISO 5, una lacuna che una SOP adeguatamente strutturata avrebbe evitato definendo protocolli di gestione degli strumenti e di introduzione sterile. La lettera di Staska Pharmaceuticals del 2025 rilevava una “convalida inadeguata della pulizia” e la necessità di una “giustificazione scientificamente supportata” dell’efficacia della pulizia dei residui e della carica batterica. L’avvertimento di Empower Pharmacy sottolinea che qualsiasi recupero microbico nella norma ISO 5 è “serio problema” e richiede un’indagine immediata, chiarendo che le SOP devono definire limiti di allarme/azione, campionamento di verifica e trigger di indagine, non semplicemente elencare le fasi di pulizia.
Le conseguenze di SOP inadeguate vanno ben oltre i risultati normativi. Le escursioni di monitoraggio ambientale sono state ricondotte alla pulizia delle stive di produzione delle forze di esecuzione, alle indagini sui batch e all'analisi approfondita delle cause principali. Se la SOP non dispone di punti di controllo di convalida o omette specifiche tecniche (ad esempio, lavaggio unidirezionale, verifica del tempo di contatto), gli investigatori faticano a determinare se l’escursione riflette un guasto del sistema di pulizia o una deviazione nell’esecuzione, prolungando le indagini ed erodendo la fiducia nell’intero programma di controllo della contaminazione.
Carenze comuni delle SOP che attivano osservazioni di conformità
Quando gli ispettori della FDA o i revisori GMP dell’UE rilasciano osservazioni sulle procedure di pulizia delle camere bianche, le carenze si concentrano attorno a diversi temi ricorrenti:
Mancanza di logica di classificazione delle aree: Le SOP generiche che applicano la stessa procedura, gli stessi strumenti e la stessa verifica alle aree ISO 5, ISO 7 e ISO 8 dimostrano una scarsa comprensione del controllo della contaminazione basato sul rischio. L'Allegato 1, Tabella 2, stabilisce i limiti di qualificazione microbica che vanno da "nessuna crescita" (grado A) a 50 CFU per piastra di contatto (grado D), ma le SOP spesso mancano di una selezione degli strumenti specifici per il grado (sterile o a basso rilascio di pelucchi), di requisiti di disinfettante (sporicida per il grado A/B rispetto a battericidi di routine per il grado C/D) o della frequenza di verifica.
Punti di controllo di convalida mancanti: Le SOP che impongono agli operatori di “lavare il pavimento” senza definire come verificare l’efficacia della pulizia, cosa costituisce una riduzione accettabile della carica batterica o quando effettuare il campionamento per il monitoraggio ambientale non lasciano prove oggettive che la pulizia raggiunga lo scopo previsto. La norma ISO 14644-5 definisce la verifica come “conferma, tramite prove oggettive, che i requisiti specificati sono stati soddisfatti”: se la SOP non fornisce criteri di misurazione, la verifica è impossibile.
Specifiche del tempo di contatto omesse: I disinfettanti richiedono un tempo di contatto umido definito per ottenere le indicazioni di uccisione etichettate. L'alcol isopropilico (IPA) al 70% è rapidamente battericida ma deve rimanere umido sulla superficie, tuttavia l'IPA evapora in pochi secondi, spesso prima di raggiungere il tempo di contatto di 1-2 minuti necessario per l'attività virucida. Le SOP che affermano "applicare disinfettante e pulire" senza specificare il tempo minimo di bagnatura, i protocolli di ribagnatura o la verifica che le superfici siano rimaste bagnate non forniscono alcuna base per presumere che sia avvenuta la disinfezione.
Nessun controllo sulla contaminazione incrociata degli strumenti: I panni riutilizzabili trasportano la carica batterica e la contaminazione da particelle provenienti dalle stanze precedenti, a meno che non vengano ritrattati secondo protocolli convalidati. Tuttavia, le SOP spesso omettono le istruzioni per il ricondizionamento, i cicli di utilizzo massimo, la verifica della sterilità o i controlli fisici che impediscono l’uso dello spazzolone a più livelli (ad esempio, codifica a colori, regole di uno spazzolone per stanza). Il risultato: un panno utilizzato in un'area di imballaggio ISO 8 entra in una stanza di supporto al riempimento ISO 6, trasportando la contaminazione che la SOP non è riuscita a prevenire.
Logica di rotazione dei disinfettanti inadeguata: L'Allegato 1 sezione 4.33 richiede "più di un disinfettante" con "diverse modalità di azione" e "uso periodico di un agente sporicida". Le POS che ruotano arbitrariamente i disinfettanti (lunedì: IPA, martedì: perossido, mercoledì: IPA) senza definire la diversità della modalità di azione, la frequenza sporicida o il monitoraggio dell’efficacia contro la flora recuperata non riescono a dimostrare un programma scientificamente giustificato.
Queste carenze sono prevenibili. Una SOP ben strutturata che definisce l'ambito in base alla classificazione ISO, integra punti di controllo di convalida con criteri di accettazione misurabili, specifica la gestione degli strumenti per eliminare la contaminazione incrociata e lega l'uso del disinfettante a tempi di contatto convalidati, trasforma la pulizia da un'attività di "massimo sforzo" in un processo controllato e verificabile che protegge la sterilità dei lotti e resiste al controllo normativo.
Panoramica della struttura SOP per il lavaggio conforme a ISO/GMP
Una SOP per il lavaggio delle camere bianche farmaceutiche non è un documento narrativo: è un quadro di controllo strutturato che guida gli operatori attraverso la preparazione pre-pulizia, l'esecuzione con verifica e la documentazione che crea una traccia di controllo. La struttura seguente riflette le migliori pratiche della norma ISO 14644-5, dell'Allegato 1 delle GMP dell'UE e delle aspettative CGMP della FDA, organizzate per supportare sia l'esecuzione da parte dell'operatore che la difesa normativa.
Componenti principali della SOP
1. Ambito & Classificazione delle aree
Definire quali camere, suite o zone disciplina la SOP, con classificazione ISO esplicita secondo ISO 14644-1 (ad esempio, Classe ISO 5, Classe 7) e i corrispondenti gradi GMP UE ove applicabile (Grado A/B/C/D). Specificare gli stati di occupazione (a riposo, operativo) ed eventuali esclusioni (ad esempio, questa POS non copre la pulizia interna delle apparecchiature o la disinfezione del soffitto). La classificazione delle aree guida ogni decisione a valle – selezione degli strumenti, requisiti di disinfettanti, frequenza di verifica – quindi l’ambiguità qui si traduce in errori di esecuzione.
Esempio di dichiarazione di ambito: "Questa SOP regola la pulizia dei pavimenti nelle anticamere di riempimento asettico di Classe ISO 6 (Grado B) e nei corridoi di vestizione di classe ISO 7 (Grado C). Le zone critiche di Classe ISO 5 (Grado A) con flusso d'aria unidirezionale sono escluse e coperte da SOP-CLEAN-005."
2. Responsabilità
Assegnare ruoli di esecuzione (operatori di produzione, servizi ambientali), revisione (supervisori della produzione), approvazione (QA) e supervisione (specialisti del controllo della contaminazione). Definire chi qualifica gli operatori, chi indaga sulle deviazioni e chi mantiene l'inventario degli strumenti e delle scorte di disinfettanti. Una chiara responsabilità previene le lacune del tipo “non è il mio lavoro” che lasciano i compiti critici ineseguiti.
3. Definizioni & Riferimenti
Definire i termini chiave che potrebbero essere interpretati erroneamente: “sterile” (sterilizzazione SAL 10⁻⁶), “basso rilascio di pelucchi” (generazione di particelle <100 particelle ≥0,5 µm per corsa), “tempo di contatto” (periodo minimo di permanenza sull'umidità per etichetta del prodotto), “sporicida” (efficace contro le spore batteriche secondo test di provocazione convalidati). Standard di riferimento (ISO 14644-1:2015, ISO 14644-5:2025, EU GMP Allegato 1 sezioni 4.33-4.36, documento CCS dell'impianto) in modo che gli auditor possano risalire ai requisiti delle normative di origine.
4. Strumenti approvati & Materiali
Elencare i sistemi di spazzolone (materiale della testina, telaio, manico), secchi o applicatori presaturati e disinfettanti per nome del prodotto, concentrazione e requisiti di tracciabilità del lotto. Specificare i requisiti di sterilità in base al grado dell'area (mop sterili irradiati con raggi gamma per il grado A/B, poliestere autoclavabile a basso rilascio di fibre per il grado C/D). Includere riferimenti alla qualificazione del fornitore e disponibilità del pacchetto di validazione (dati sulla generazione delle particelle, matrici di compatibilità chimica, certificati di sterilizzazione). Questa sezione risponde alla domanda dell'operatore: "Cosa posso usare?"
5. Preparazioni pre-lavaggio
Dettagliare i requisiti dell'abbigliamento, i protocolli di introduzione del materiale (disinfezione dell'esterno della confezione del mop prima dell'ingresso nella camera di equilibrio), controlli di integrità dello strumento (confezione sterile sigillata intatta, nessun danno visibile alle testine del mop) e preparazione del disinfettante (verifica della diluizione, sterilità per il grado A/B, data di scadenza). È qui che viene prevenuta o introdotta la contaminazione incrociata: rigorosi controlli preliminari individuano i problemi prima che entrino nella camera bianca.
6. Procedura dettagliata
Fornire istruzioni sequenziali eseguibili dall'operatore relative all'impostazione della stanza (verifica HEPA, percorso del flusso del materiale), sequenza di pulizia (dall'alto verso il basso per le pareti, da lontano all'uscita per i pavimenti, movimenti unidirezionali allineati al flusso d'aria), verifica del tempo di contatto (la superficie rimane visibilmente bagnata per la durata indicata sull'etichetta) e controlli post-lavaggio (nessun ristagno, nessuna striscia di residui). Utilizza passaggi numerati, non paragrafi. Ogni fase dovrebbe avere una singola azione e, se pertinente, un criterio di accettazione.
7. Verifica & Documentazione
Definire cosa devono registrare gli operatori (data, ora, area, lotto di disinfettante, iniziali dell'operatore, eventuali deviazioni), come si verificano i collegamenti al monitoraggio ambientale (ad esempio, "Il QA raccoglierà le piastre di sedimentazione nell'area lavata entro 30 minuti dopo la pulizia") e i fattori scatenanti dell'indagine (residui visibili, tempo di contatto non raggiunto, guasto dell'integrità dello strumento). La documentazione è la prova oggettiva richiesta dalla norma ISO 14644-5 per la verifica: senza di essa, la pulizia non è verificabile.
8. Sicurezza & Precauzioni
Affrontare i rischi chimici (infiammabilità dell'IPA, avvertenze sul perossido ossidante), i rischi di scivolamento derivanti da pavimenti bagnati e i contatti di emergenza. Questa sezione protegge gli operatori e dimostra la due diligence nella gestione del rischio.
9. Appendici & Riferimenti
Allegare tabelle dei tempi di contatto con i disinfettanti, diagrammi di flusso per il ricondizionamento degli strumenti (per sistemi riutilizzabili), schemi di codifica a colori che impediscono l'uso tra diversi livelli e collegamenti alle procedure operative standard correlate (camici, smaltimento dei rifiuti, monitoraggio ambientale). Le appendici mantengono snella la procedura principale fornendo agli operatori dettagli di riferimento rapido quando necessario.
Questa struttura trasforma una vaga istruzione di “pulire il pavimento” in un processo controllato con input definiti (strumenti qualificati, disinfettanti convalidati), esecuzione controllata (tecnica unidirezionale, tempo di contatto verificato) e output misurabili (completamento documentato, verifica EM entro criteri di accettazione). Gli enti regolatori si aspettano questo livello di rigore nella produzione farmaceutica: fornirlo attraverso SOP strutturate non è facoltativo.

Figura 1: Diagramma di flusso della struttura SOP conforme a ISO/GMP. Questo quadro modulare garantisce la difendibilità normativa definendo l'ambito in base alla classificazione ISO, integrando punti di controllo di convalida e creando procedure eseguibili dall'operatore con risultati misurabili.
Flusso di lavoro passo passo per il lavaggio delle camere bianche (versione ad alta conformità)
Il nucleo operativo di una SOP per il lavaggio di camere bianche è la procedura passo passo eseguita dagli operatori. Questo flusso di lavoro integra la logica di classificazione ISO, i requisiti degli strumenti sterili dell'Allegato 1, la verifica del tempo di contatto e i controlli della contaminazione incrociata in una sequenza strutturata che trasforma i mandati normativi in pratica quotidiana.
1. Preparazione pre-pulizia
Prima che qualsiasi spazzolone entri in un'area classificata, gli operatori devono completare i controlli relativi all'abbigliamento, alla qualificazione degli strumenti e all'idoneità ambientale.
Qualifica di vestizione: Il personale deve completare l'abbigliamento per camera bianca specifico per la struttura in base alle SOP del sito, con riqualificazione annuale per l'accesso di Grado A/B (l'Allegato 1 richiede una valutazione "almeno una volta all'anno" per il personale dell'area asettica). L'integrità del camice, la tecnica di indossare i guanti e la vestibilità del cappuccio/occhiali devono soddisfare i criteri di accettazione prima di procedere. Gli operatori con difetti visibili dell'abito (guanti strappati, capelli scoperti) sono immediatamente squalificati dall'ingresso.
Verifica dell'integrità del panno: Rimuovere il panno dall'imballaggio e verificare eventuali danni. Per i panni monouso sterili irradiati con raggi gamma (grado A/B), verificare che il sigillo della confezione sia intatto, che l'indicatore di radiazione mostri un cambiamento di colore "superato" e che la data di scadenza della sterilità sia valida. Per i panni riutilizzabili autoclavabili, verificare che l'indicatore del nastro dell'autoclave indichi "sterile" e che la data di sterilizzazione rientri nel tempo di attesa (in genere 7-30 giorni a seconda della confezione). Qualsiasi indicatore di sterilità compromessa fa scattare il rifiuto del panno e la documentazione come deviazione.
Preparazione e verifica del disinfettante: Per le aree di Grado A/B, i disinfettanti devono essere sterili prima dell'uso (Allegato 1 sezione 4.34). Utilizzare un disinfettante sterile preparato commercialmente con certificato di analisi tracciato per lotto oppure preparare le diluizioni in modo asettico utilizzando acqua sterile e una tecnica asettica convalidata. Verificare la concentrazione utilizzando le tabelle di diluizione riportate sull'etichetta o il rifrattometro (per IPA). Registrare il numero di lotto, la data/ora di preparazione, la scadenza (in genere 24 ore per le soluzioni preparate in struttura) e le iniziali dell'operatore. Per il Grado C/D, i disinfettanti non sterili di grado farmaceutico sono accettabili se validati efficaci.
Protocollo di introduzione del materiale: Disinfettare le superfici esterne di tutti i materiali che entrano nella camera bianca (imballaggi di stracci, bottiglie di disinfettante) all'ingresso della camera di equilibrio utilizzando la SOP di disinfezione del trasferimento della struttura. Consentire il tempo di contatto per SOP prima del trasferimento attraverso la camera di equilibrio. Ciò impedisce l'introduzione di contaminazioni esterne sugli imballaggi che successivamente entreranno in contatto con le superfici delle camere bianche.
2. Allestimento della sala e verifica ambientale
Conferma del flusso d'aria HEPA: Prima di iniziare la pulizia, verificare che il sistema HEPA della stanza sia in funzione. Controllare che i manometri differenziali mostrino una pressione positiva relativa alle aree adiacenti di grado inferiore (tipico: +10-15 Pa Grado B→C, +15-20 Pa Grado A→B). Per le aree con flusso d'aria unidirezionale (Grado A), confermare visivamente il primo flusso d'aria utilizzando il metodo di visualizzazione del flusso d'aria della struttura (ad esempio, streamer di particelle approvati, studi sul fumo della struttura). Se il flusso d'aria è compromesso, avvisare il supervisore e ritardare la pulizia finché non viene risolta; la pulizia senza un sistema HEPA funzionale rischia di accumulare contaminazione.
Creazione del percorso del flusso del materiale: libera un percorso definito dalla camera di equilibrio all'angolo più lontano della stanza. La pulizia procede da questo angolo lontano verso l'uscita, garantendo che gli operatori non camminino mai sui pavimenti appena lavati e non depositino nuovamente la contaminazione. Rimuovere le attrezzature mobili o i materiali che ostruiscono il percorso dello spazzolone. Nelle linee di riempimento di Grado A, coordinarsi con la produzione per garantire che le operazioni asettiche siano completate e che la linea sia nello stato "a riposo" prima che inizi il lavaggio.
Sequenza di pre-asciugatura e quindi di lavaggio: L'Allegato 1 e le migliori pratiche GMP richiedono la pulizia prima della disinfezione perché detriti e residui inibiscono il contatto con il disinfettante. Per pareti e superfici verticali, pulire prima utilizzando salviette approvate a basso rilascio di pelucchi per rimuovere le particelle visibili, quindi applicare un panno disinfettante. Per i pavimenti con detriti visibili, eseguire un pre-aspirazione utilizzando un aspirapolvere per camere bianche con filtro HEPA o un pre-aspirazione delle zone ad alto traffico prima della pulizia a umido. Questo approccio in due fasi garantisce che il disinfettante venga a contatto con una superficie pulita, massimizzandone l'efficacia.
3. Sequenza di lavaggio: unidirezionale, sovrapposta, percorso di uscita allineato
Principio dall'alto verso il basso, pulito-sporco: Pulire sempre le superfici superiori prima di quelle inferiori. Per le pareti, iniziare dalla giunzione del soffitto e pulire verso il basso con movimenti verticali. Per i pavimenti, iniziare dall'angolo più lontano e pulito e procedere verso l'uscita (in genere la camera di equilibrio o l'ingresso dell'area di vestizione). Non pulire mai le aree pulite da quelle sporche: ciò provoca contaminazione incrociata e viola la logica di controllo della contaminazione.
Tecnica del colpo unidirezionale: utilizzare tratti diritti e paralleli solo in una direzione. Tirare il panno verso di sé con un movimento fluido e controllato, sollevarlo alla fine della corsa, riposizionarlo per la passata successiva e ripetere. Non eseguire movimenti avanti e indietro o a forma di 8: questi ridistribuiscono la contaminazione anziché rimuoverla. Ogni tratto dovrebbe sovrapporsi al precedente del 10-25% per garantire che non vi siano spazi vuoti nella copertura.
Allineamento del flusso d'aria per il Grado A/B: Nelle aree con flusso d'aria unidirezionale, allineare le passate del panno con la direzione del flusso d'aria HEPA, quando possibile. Lo straccio perpendicolare alla prima aria può interrompere il flusso laminare e aumentare temporaneamente il numero di particelle nella zona critica. Sebbene alcune interruzioni siano inevitabili durante la pulizia, l'allineamento con il flusso d'aria riduce al minimo le turbolenze e supporta un ritorno più rapido alla classificazione.
Verifica del tempo di contatto (punto critico di conformità): Dopo aver applicato il disinfettante, la superficie deve rimanere visibilmente bagnata per il tempo di contatto indicato sull'etichetta. È qui che la maggior parte delle SOP fallisce nell'esecuzione. L'alcol isopropilico (70% IPA) evapora in 30-60 secondi ma richiede 1-2 minuti per la completa attività virucida. Le formulazioni di perossido di idrogeno specificano tipicamente un contatto di 1-5 minuti a seconda della concentrazione. Gli operatori devono applicare un volume sufficiente per mantenere l'umidità durante il periodo di contatto oppure bagnare nuovamente le superfici che si asciugano prematuramente. Documentare il raggiungimento del tempo di contatto: "La superficie è rimasta visibilmente bagnata per 3 minuti secondo il requisito SOP-CHEM-012."
Regola di uno straccio per stanza (prevenzione della contaminazione incrociata): I panni usa e getta vengono scartati dopo aver completato una stanza e mai riutilizzati. I panni riutilizzabili devono essere raccolti per il ricondizionamento dopo ogni stanza e mai trasportati da un'area all'altra senza una sterilizzazione convalidata. Se si utilizzano sistemi con codice colore, applicare una rigorosa separazione dei gradi: panni blu solo per il grado C, gialli solo per il grado B, rossi solo per il grado A. L'uso dello spazzolone a livello trasversale è una deviazione critica che richiede un'indagine.
4. Documentazione del tempo di contatto con il disinfettante
Il tempo di contatto non è facoltativo: è un parametro convalidato che deve essere registrato e verificato. Le POS devono specificare:
- Nome del prodotto e numero di lotto del disinfettante utilizzato
- Tempo di contatto indicato in base alle istruzioni per l'uso del produttore o alla convalida della struttura (ad esempio, "3 minuti di contatto bagnato")
- Metodo di verifica: conferma visiva dell'umidità mantenuta per tutto il periodo o documentazione del timer
- Protocollo di riumidificazione se la superficie si asciuga prematuramente: "Se la superficie si asciuga prima che sia trascorso il tempo di contatto, applicare nuovamente il disinfettante e riavviare il timer"
Le lettere di avvertimento della FDA citano specificamente il tempo di contatto insufficiente come una lacuna di convalida ricorrente. La lettera di Staska del 2025 sottolineava la necessità di una “giustificazione scientificamente supportata” dell’efficacia della pulizia, che dipende dal raggiungimento di condizioni di esposizione convalidate. Una POS che omette il tempo di contatto non lascia alcuna base per affermare che sia avvenuta la disinfezione.
5. Percorso di smaltimento o ricondizionamento
Smaltimento del mocio usa e getta: Dopo l'uso, i panni monouso vengono immediatamente collocati negli appositi contenitori per rifiuti all'interno della camera bianca (tipicamente rivestiti con sacchetti per autoclave per l'inattivazione della carica batterica prima della rimozione). Registrare lo smaltimento nel registro delle pulizie: "Mop n. 2025-04-18-001 utilizzato nella stanza 204 (ISO 6), scartato secondo SOP-WASTE-003." Il percorso di smaltimento offre un rischio di contaminazione incrociata pari a zero ed elimina l'onere della convalida del ritrattamento, un vantaggio chiave per le aree ad alto rischio.
Ricondizionamento del mop riutilizzabile: Raccogliere i panni riutilizzabili usati negli appositi contenitori per gli attrezzi sporchi, separati per grado. Trasporto alla lavanderia convalidata o all'area di ritrattamento in loco in base alla SOP del flusso di materiale. Il ricondizionamento deve includere:
- Ciclo di lavaggio convalidato (temperatura, detersivo, cicli di risciacquo) qualificato per rimuovere la carica batterica e le particelle senza degradare il materiale del panno
- Ispezione per usura, strappi o perdita di prestazioni a basso rilascio di pelucchi; rifiutare i panni che superano il numero massimo di cicli (ad esempio, 50-100 cicli di bucato a seconda del materiale)
- Sterilizzazione (autoclavaggio a 121°C per 30 minuti o irradiazione gamma) con verifica della sterilità secondo protocollo di validazione
- Confezione sterile con data di scadenza e tracciabilità del lotto
Ignorare i limiti del conteggio dei cicli è una pratica ad alto rischio. I panni si degradano con ripetuti lavaggi e sterilizzazioni, rilasciando progressivamente più particelle e perdendo capacità di assorbimento. Le SOP devono definire i criteri di rifiuto e documentare il monitoraggio del conteggio dei cicli.

Figura 2: Flusso di lavoro di pulizia per camere bianche ad alta conformità. Ogni fase prevede checkpoint di verifica che trasformano i mandati normativi in passaggi eseguibili dall'operatore con prove oggettive di conformità.
Requisiti di convalida per le SOP di lavaggio
Una POS di pulizia non convalidata è, da un punto di vista normativo, non controllata. L'allegato 1 delle GMP UE, sezione 4.22, richiede che la "pulizia prima della disinfezione" sia convalidata e che i processi di disinfezione siano convalidati "nelle specifiche modalità d'uso". La norma ISO 14644-5 richiede la verifica attraverso prove oggettive. Le lettere di avvertimento della FDA citano “convalida inadeguata della pulizia” quando le strutture non possono dimostrare, con dati, che la pulizia raggiunga il controllo della contaminazione previsto. Questa sezione definisce i punti di controllo di convalida che trasformano una procedura scritta in un sistema di pulizia qualificato e difendibile.
1. Convalida delle particelle (conformità ISO 14644-1)
Il lavaggio non deve violare i limiti di classificazione della stanza durante l'esecuzione o impedire il tempestivo recupero della classificazione a riposo dopo la pulizia.
Approccio di validazione: Condurre il monitoraggio delle particelle durante le operazioni di pulizia nel caso peggiore (superficie massima del pavimento, operatore rappresentativo, disinfettante di routine) utilizzando contatori ottici di particelle calibrati posizionati secondo i piani di campionamento ISO 14644-1. Misurare la concentrazione delle particelle a ≥ 0,5 µm e ≥ 5,0 µm prima dello straccio, durante lo straccio (disturbo di picco) e a intervalli definiti dopo lo straccio fino al ritorno ai valori di base.
Criteri di accettazione per classe ISO:
- Classe ISO 5: Il conteggio delle particelle deve ritornare a ≤3.520 particelle/m³ (≥0,5 µm) entro 15-20 minuti dopo lo lavaggio. I picchi transitori durante il lavaggio non devono superare il limite di classificazione pari a 2x. Un superamento prolungato attiva l'indagine sulla generazione di particelle dello spazzolone, sulla tecnica dell'operatore o sull'interruzione del flusso d'aria.
- Classe ISO 6: Ritorna a ≤35.200 particelle/m³ (≥0,5 µm) entro 20 minuti. Picco durante il lavaggio <Limite 2,5x.
- Classe ISO 7: Ritorno a ≤352.000 particelle/m³ (≥0,5 µm) entro 20 minuti. Picco durante il lavaggio <Limite 3x.
- Classe ISO 8: Ritorno a ≤3.520.000 particelle/m³ (≥0,5 µm) entro 20 minuti.
Perché i panni riutilizzabili non superano più spesso la validazione delle particelle: I panni riutilizzabili lavati rilasciano progressivamente più particelle man mano che il tessuto si degrada con cicli ripetuti di lavaggio/sterilizzazione. Anche con un ritrattamento convalidato, la variabilità da ciclo a ciclo introduce il rischio di generazione di particelle. I panni usa e getta offrono prestazioni costanti e qualificate in termini di particelle perché ogni utilizzo inizia con una testina nuova e convalidata in fabbrica.
2. Convalida della carica batterica
La pulizia e la disinfezione devono ottenere una riduzione misurabile della carica batterica. L'Allegato 1 Tabella 2 definisce i limiti di qualificazione microbica per grado; il monitoraggio ambientale di routine verifica il controllo continuo.
Tendenza della carica batterica al basale: Stabilire un livello di riferimento della carica batterica pre-pulizia attraverso il campionamento EM di routine (piastre di contatto, tamponi) raccolto da superfici rappresentative del pavimento prima dello lavaggio programmato. Linea di base dell'andamento su 3-6 mesi per comprendere i livelli tipici di contaminazione in base all'area e all'ora del giorno. I campioni post-pulizia raccolti 30-60 minuti dopo il lavaggio (consentendo il tempo di contatto con il disinfettante e l'evaporazione) dimostrano una riduzione della carica batterica.
Criteri di accettazione (risultati EM post-pulizia):
- Grado A: Nessuna crescita (0 CFU) su piastre a contatto o tamponi
- Grado B: ≤5 CFU per piastra di contatto
- Grado C: ≤25 CFU per piastra di contatto
- Grado D: ≤50 CFU per piastra di contatto
Il mancato rispetto di questi limiti avvia l'indagine: è stato raggiunto il tempo di contatto? Il disinfettante è ancora efficace contro la flora ripristinata? Il ritrattamento del mop è adeguato (per i riutilizzabili)? La tecnica dell’operatore introduce contaminazione?
Perché i panni riutilizzabili comportano un rischio maggiore di carica batterica: I panni riutilizzabili possono ospitare carica batterica residua negli interstizi del tessuto se il lavaggio è inadeguato o se i panni vengono conservati umidi tra un utilizzo e l'altro. Anche il ritrattamento convalidato può fallire se i parametri del ciclo si discostano (ad esempio, la temperatura di lavaggio scende al di sotto del setpoint di convalida). I panni sterili monouso eliminano questo rischio: ogni utilizzo è sterile SAL 10⁻⁶.
3. Convalida del disinfettante
I disinfettanti devono essere validati efficaci contro la carica batterica della struttura, su superfici rappresentative, nella modalità di utilizzo (lavaggio), con tempo di contatto convalidato.
Logica di rotazione (modalità di azione diversità): L’Allegato 1 richiede “più di un disinfettante” con “diverse modalità di azione” e “uso periodico di un agente sporicida”. Una rotazione conforme potrebbe essere:
- Settimana 1-3: Alcool isopropilico al 70% (alcol, battericida/virucida)
- Settimana 4: Perossido di idrogeno accelerato allo 0,5% (ossidante, battericida/fungicida/virucida)
- Mensile: Ipoclorito di sodio 5000 ppm (ossidante, sporicida) o acido peracetico (sporicida)
Questa rotazione impedisce alla flora di adattarsi a un singolo disinfettante e affronta gli organismi che formano spore (ad es. Bacillo spp.) che resistono all'alcol.
Protocollo di verifica del tempo di contatto: Convalidare che gli operatori possano mantenere il tempo di contatto bagnato etichettato nelle condizioni di utilizzo effettive. Utilizzare l'osservazione visiva, il monitoraggio della temperatura superficiale (per prodotti chimici sensibili alla temperatura) o traccianti coloranti per confermare la copertura e il tempo di permanenza. Se l'alcool evapora prima che sia trascorso il tempo di contatto, modificare il volume o la tecnica di applicazione oppure prendere in considerazione mop pre-saturati che forniscono un volume di soluzione controllato.
Convalida della compatibilità (fondamentale per i mop riutilizzabili): Alcune combinazioni di materiale disinfettante riducono le prestazioni del panno. I composti di ammonio quaternario (quat) si adsorbono sulle fibre di cellulosa e perdono attività; sono necessarie teste per spazzoloni in poliestere. La candeggina degrada alcuni polimeri compatibili con l'autoclave nel corso di esposizioni ripetute. La convalida deve dimostrare che i materiali approvati per i mop tollerano tutti i disinfettanti nel programma di rotazione senza perdita di proprietà di basso rilascio di pelucchi o di integrità strutturale.
4. Qualificazione dell'Operatore (OQ)
L'efficacia della pulizia dipende dalla tecnica dell'operatore. L'Allegato 1 richiede formazione e rivalutazione periodica; La norma ISO 14644-5 richiede la qualificazione del personale integrata nell'OCP.
Qualificazione iniziale: Gli operatori completano la formazione pratica sulle fasi SOP, l'abbigliamento, la manipolazione dello spazzolone, la tecnica della corsa unidirezionale, la verifica del tempo di contatto e la documentazione. La qualifica include la valutazione delle prestazioni osservate: l'allenatore osserva l'operatore eseguire il ciclo completo di lavaggio e verifica la tecnica rispetto alla lista di controllo (colpi unidirezionali, corretta sovrapposizione, tempo di contatto raggiunto, regola di uno straccio per stanza seguita). Solo gli operatori con abilitazione “pass” sono autorizzati a pulire le aree classificate senza sorveglianza.
Riqualificazione annuale delle competenze: L'Allegato 1 richiede una rivalutazione “almeno una volta all'anno” per il personale di Grado A/B. La riqualificazione comprende una valutazione scritta (conoscenza della SOP, tempi di contatto con il disinfettante, segnalazione delle deviazioni) e le prestazioni osservate. Qualsiasi operatore collegato all'escursione EM o alla deviazione dalla SOP viene sottoposto a riqualificazione e riqualificazione immediata prima di riprendere le attività di pulizia.
Registrazione video come prova: Alcune strutture registrano le sessioni di qualificazione degli operatori, creando prove oggettive per gli audit. Le sessioni registrate dimostrano la tecnica corretta e forniscono materiale di formazione per i nuovi assunti. Il video non è richiesto dalla normativa ma offre una solida documentazione attestante che gli operatori sono qualificati secondo le aspettative GMP.
5. Convalida degli strumenti: due percorsi con oneri drammaticamente diversi
La scelta tra mop usa e getta e riutilizzabili crea carichi di lavoro di convalida molto diversi.
Percorso di validazione dei mop monouso (semplificato):
- Qualificazione dei materiali: Il fornitore fornisce dati sulla generazione di particelle (secondo ISO 14644-14 o equivalente), matrici di compatibilità chimica e certificati di sterilità (per mop irradiati con raggi gamma: rapporti di convalida SAL 10⁻⁶). La struttura esamina i dati, qualifica il programma di qualità fornitore per fornitore e approva il materiale per l'uso.
- Verifica della sterilità: Per i prodotti monouso sterili, i test periodici di sterilità dei lotti in entrata (ad esempio, trimestralmente o secondo un piano di campionamento basato sul rischio) confermano che il processo di sterilizzazione del fornitore rimane sotto controllo. I certificati di analisi (CoA) documentano i risultati dei test di sterilità.
- Monitoraggio continuo: Gli studi di validazione delle particelle confermano che i materiali usa e getta non contribuiscono alle escursioni delle particelle durante l'uso. Poiché ogni panno è monouso, il conteggio dei cicli e la convalida del ritrattamento vengono eliminati.
Onere totale della convalida: qualificazione del materiale + verifica della sterilità + studi sull'uso delle particelle.
Percorso di convalida del mocio riutilizzabile (complesso):
- Qualificazione dei materiali: Uguale al monouso (generazione di particelle, compatibilità chimica)
- Riciclaggio IQ/OQ/PQ: Convalidare il processo di lavaggio (Qualificazione dell'installazione delle attrezzature della lavanderia, Qualificazione operativa dei parametri del ciclo di lavaggio, Qualificazione delle prestazioni che dimostra la rimozione della carica batterica e il mantenimento delle prestazioni delle particelle). Ciò richiede test di sfida con mop sporchi artificialmente, studi sul recupero della carica batterica e test sulla generazione di particelle prima e dopo il lavaggio.
- Sterilizzazione IQ/OQ/PQ: Convalida del ciclo dell'autoclave o del processo di irradiazione gamma, comprese le verifiche degli indicatori biologici, la mappatura della temperatura e la verifica dei test di sterilità.
- Limiti del conteggio dei cicli: Definire i cicli massimi di bucato/sterilizzazione prima del rifiuto del panno (ad esempio, 50 cicli). Richiede studi sull'invecchiamento accelerato che dimostrino che la generazione di particelle e l'assorbenza rimangono accettabili fino alla fine del ciclo di vita qualificato.
- Garanzia di sterilità: test periodici di sterilità dei panni trattati, tempo di permanenza sterile convalidato post-sterilizzazione (ad esempio, 30 giorni in confezione sigillata) e data di scadenza.
- Tracciamento del ritrattamento: Implementare un sistema per tenere traccia del conteggio dei singoli cicli di pulizia (ad esempio, tag RFID, etichette con codici a barre), imporre il ritiro ai cicli massimi e documentare i record dei lotti di rielaborazione secondo GMP.
Onere totale della convalida: qualificazione del materiale + IQ/OQ/PQ di riciclaggio + IQ/OQ/PQ di sterilizzazione + studi sul conteggio dei cicli + garanzia di sterilità + convalida del sistema di tracciamento.
Conclusione: i sistemi di scopa monouso riducono l'onere di convalida del 60-70% rispetto ai riutilizzabili, eliminano il rischio di contaminazione correlato al ritrattamento e forniscono prestazioni prevedibili delle particelle. Per le aree ad alto rischio ISO 5-6, i vantaggi del risparmio di convalida e del controllo della contaminazione favoriscono fortemente i prodotti monouso.
Cosa fare & Da non fare nel lavaggio Pharma Cleanzone
Cosa fare & Da non fare nel lavaggio Pharma Cleanzone
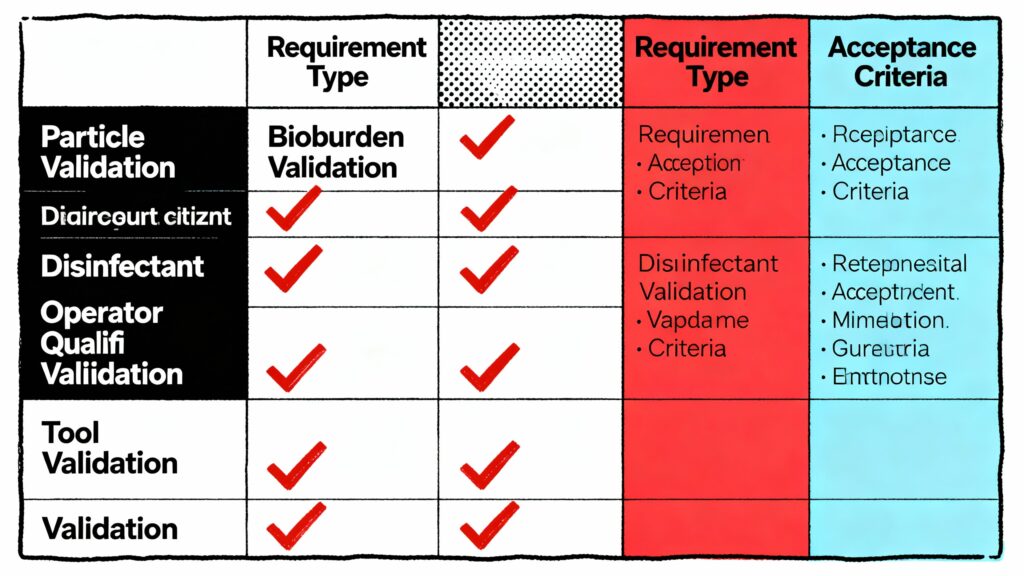
Figura 3: Matrice dei requisiti di convalida per le SOP per la pulizia delle camere bianche. Cinque punti di controllo di convalida (particelle, carica batterica, disinfettante, qualifica dell'operatore, convalida dello strumento) con criteri di accettazione espliciti dimostrano che la pulizia raggiunge il controllo della contaminazione previsto secondo i requisiti dell'Allegato 1 e della norma ISO 14644-5.
Cosa fare (pratiche corrette)
Utilizzare panni sterili e sigillati nelle aree di grado A/B: L'Allegato 1 sezione 4.34 richiede disinfettanti sterili e strumenti di pulizia nelle zone asettiche. Per il Grado A/B, utilizzare spazzoloni monouso sterili irradiati con raggi gamma (SAL 10⁻⁶) forniti in confezioni sigillate con indicatori di radiazione o riutilizzabili autoclavabili sterilizzati immediatamente prima dell'uso con garanzia di sterilità convalidata. I panni non sterili nelle aree asettiche rappresentano una violazione critica delle GMP.
Se possibile, pulire sempre nella direzione del flusso d'aria unidirezionale: Il flusso d'aria unidirezionale (laminare) di Grado A spinge le particelle lontano dalle superfici critiche. Il lavaggio perpendicolare al flusso d'aria crea turbolenza che può sospendere temporaneamente le particelle sul prodotto esposto. Allinea le passate del panno con la direzione del flusso d'aria per ridurre al minimo i disagi e favorire una più rapida rimozione delle particelle dopo la pulizia.
Tieni traccia del lotto, della scadenza e del tempo di contatto del disinfettante nei registri di pulizia: Ogni evento di pulizia deve documentare il numero di lotto del disinfettante, la data di preparazione (per le soluzioni preparate in struttura), la data/ora di scadenza e il tempo di contatto raggiunto. Questa documentazione fornisce prove oggettive del rispetto dei parametri di pulizia convalidati: senza di essa, non è possibile dimostrare la conformità durante indagini o audit.
Implementare e far rispettare la regola dello spazzolone per stanza: La contaminazione incrociata dovuta ai panni utilizzati in più stanze senza ritrattamento è una delle principali cause delle escursioni di monitoraggio ambientale. I panni usa e getta vengono gettati dopo ogni stanza. I panni riutilizzabili devono essere codificati a colori in base al grado dell'area (ad esempio, rosso solo per il grado A, giallo solo per il grado B, blu solo per il grado C) e raccolti per il ricondizionamento convalidato dopo l'uso in una singola stanza. Non trasportare mai uno spazzolone usato da una stanza all'altra.
Verificare che la superficie rimanga bagnata per il tempo di contatto indicato sull'etichetta: L'efficacia disinfettante dipende dal raggiungimento del tempo di contatto specificato dal produttore con la superficie visibilmente bagnata. Per gli agenti a evaporazione rapida come l'alcol isopropilico, ciò potrebbe richiedere la riapplicazione o un volume iniziale più elevato per mantenere l'umidità durante l'intero periodo di contatto. Gli operatori devono verificare visivamente l'umidità ovunque: se la superficie si asciuga presto, bagnarla nuovamente e riavviare il timer del tempo di contatto.
Completare la qualificazione dell'abbigliamento prima di accedere alle aree classificate: Tutto il personale che accede alle aree ISO 5-8 deve completare le SOP relative all'abbigliamento per sito e superare le valutazioni di qualificazione dell'abbigliamento. Per il Grado A/B l'Allegato 1 prevede la riqualificazione annuale “almeno una volta all'anno” con valutazione visiva e microbica. Il personale non qualificato introduce un rischio di contaminazione che il lavaggio non può risolvere.
Da non fare (pratiche vietate)
Non riutilizzare i panni tra gradi ISO o stanze adiacenti: L'utilizzo dello stesso panno in confezione ISO 8 e poi del supporto di riempimento ISO 6 senza ritrattamento provoca la contaminazione incrociata delle aree più pulite con carica batterica e particelle provenienti da zone di qualità inferiore. Questa pratica viola i principi di controllo della contaminazione ed è un’osservazione comune della FDA/EMA. Ciascun grado di area richiede panni dedicati o un ritrattamento convalidato tra un utilizzo e l'altro.
Non utilizzare movimenti a forma di 8 o di pulizia avanti e indietro: La Figura 8 e i movimenti avanti e indietro ridistribuiscono la contaminazione anziché rimuoverla, diffondono il disinfettante in modo non uniforme e creano zone umide sovrapposte dove il tempo di contatto è imprevedibile. Utilizzare passate unidirezionali, diritte e parallele con una sovrapposizione del 10-25% e sollevare il panno alla fine di ogni passata prima di riposizionarlo.
Non immergere due volte il panno nel secchio dopo il contatto con il pavimento: Una volta che lo spazzolone tocca il pavimento, è contaminato. Immergerlo nuovamente nel secchio del disinfettante contamina la soluzione, riducendone l'efficacia per le applicazioni successive. Utilizzare mop presaturati monouso o implementare sistemi a secchio convalidati con strizzatori che separano la soluzione pulita da quella usata. Per i sistemi riutilizzabili, cambiare la soluzione disinfettante dopo ogni stanza o quando visibilmente sporca.
Non utilizzare processi di lavanderia non convalidati per i panni riutilizzabili: L'invio di panni per camere bianche a una lavanderia commerciale senza una rimozione convalidata della carica batterica, test sulla generazione di particelle e controlli dei parametri del ciclo (temperatura di lavaggio, tipo di detersivo, qualità del risciacquo) rischia di introdurre contaminazione in aree classificate. I panni riutilizzabili richiedono un lavaggio qualificato IQ/OQ/PQ con registri dei lotti documentati, monitoraggio del conteggio dei cicli e verifica periodica della sterilità. La lavanderia non convalidata non è controllata e non è conforme alle GMP.
Non saltare la rotazione dei disinfettanti né omettere sporicidi periodici: L'utilizzo di un solo disinfettante (ad esempio, esclusivamente IPA al 70%) consente alla flora di adattarsi e ai formatori di spore di persistere. L’Allegato 1 richiede “più di un disinfettante” con “diverse modalità di azione” e “l’uso periodico di un agente sporicida”. Un programma conforme ruota almeno due disinfettanti (ad esempio, alcool + ossidante) e include cicli sporicidi mensili o trimestrali (ipoclorito di sodio, acido peracetico o perossido di idrogeno a concentrazione sporicida). Il monitoraggio della sensibilità dell'isolato ambientale ai disinfettanti verifica l'efficacia del programma.
Non pulire durante le operazioni asettiche attive nel Grado A: Il lavaggio genera disturbi transitori di particelle e richiede la presenza/movimento dell'operatore che può interrompere il flusso d'aria laminare sul prodotto sterile esposto. Pianifica il lavaggio durante i tempi di inattività della linea, tra un lotto e l'altro o durante i periodi di qualificazione "a riposo" convalidati. Il lavaggio con lo straccio durante le operazioni di riempimento attive di Grado A è vietato in base alle aspettative di controllo della contaminazione dell'Allegato 1.
Non superare il numero massimo di cicli per i mop riutilizzabili: I panni si degradano con ripetuti lavaggi e sterilizzazioni. Le fibre del tessuto si sfilacciano, i bordi sigillati si delaminano e la generazione di particelle aumenta. La convalida definisce i cicli massimi qualificati (ad esempio, 50-100 cicli). Superare questo limite senza riconvalida significa che il mop non funziona più secondo le specifiche. Implementare sistemi di tracciabilità (codici a barre, tag RFID) per far rispettare i limiti di ciclo e ritirare i panni a fine vita.
Non introdurre disinfettante non sterile nelle aree di Grado A/B: L'Allegato 1 sezione 4.34 richiede che i disinfettanti utilizzati di Grado A/B siano sterili. Le diluizioni preparate in struttura devono essere effettuate in modo asettico utilizzando acqua sterile, convalidate per la sterilità e assegnati limiti di tempo di attesa (tipicamente 24 ore). I disinfettanti sterili forniti in commercio devono essere qualificati dal fornitore con certificati di analisi. L’uso di disinfettanti non sterili in zone asettiche introduce carica batterica vitale che compromette l’intero programma di controllo della contaminazione.
Rischi nascosti di contaminazione incrociata (punti di consapevolezza)
Gli operatori possono inconsapevolmente introdurre contaminazione attraverso pratiche che appaiono ragionevoli ma violano i principi GMP:
- Conservazione dei panni riutilizzabili bagnati: I panni conservati umidi tra un utilizzo e l'altro diventano incubatori per la carica batterica. Tutti i mop riutilizzabili devono essere lavati, sterilizzati e confezionati immediatamente dopo l'uso, non appesi ad "asciugare" per un successivo riutilizzo.
- Utilizzo di salviette a base di cellulosa con disinfettanti QAC: I composti di ammonio quaternario si adsorbono sulle fibre di cellulosa e perdono attività antimicrobica. Utilizzare panni/mop in poliestere o altri sintetici a basso rilascio di pelucchi con QAC.
- Lavaggio immediato dopo la sostituzione del filtro HEPA senza verifica del flusso d'aria: Le modifiche al filtro possono introdurre particolato. Verificare che la stanza ritorni alla classificazione (monitoraggio del conteggio delle particelle) prima di riprendere la pulizia di routine per evitare la diffusione di detriti di installazione.
- Ignorare i danni visibili del mop: Testine strappate, bordi sfilacciati o confezioni sterili danneggiate compromettono il controllo delle particelle e la garanzia di sterilità. Rifiutare immediatamente i panni danneggiati e documentarli come deviazione.
Codificando queste pratiche nella formazione SOP e nelle valutazioni delle competenze, le strutture riducono la variabilità dell'esecuzione e prevengono eventi di contaminazione ricorrenti riconducibili a errori dell'operatore.
Strumenti consigliati per scrivere una SOP (conforme a GMP)

Figura 4: Cosa fare e cosa non fare nella pulizia delle camere bianche farmaceutiche. A sinistra: pratiche corrette (passate unidirezionali, uno spazzolone per stanza, verifica del tempo di contatto). A destra: pratiche vietate che innescano osservazioni della FDA ed escursioni di monitoraggio ambientale (movimento a 8, riutilizzo trasversale, double-dipping).
Una volta definiti la struttura della SOP, il flusso di lavoro e i requisiti di convalida, la selezione dello strumento diventa la decisione pratica che determina se la SOP è eseguibile e difendibile. Gli “strumenti approvati & materiali" della POS dovrebbe fare riferimento a prodotti che soddisfano le aspettative normative, sono dotati di pacchetti di supporto per la convalida e corrispondono alla classificazione ISO e al modello operativo della vostra struttura.
Criteri di selezione degli strumenti per i mop con riferimento a SOP
Percorso di sterilità per Grado A/B: Per le aree asettiche, i panni devono essere sterili prima dell'uso (requisito dell'Allegato 1). Due percorsi soddisfano questo:
- Mop sterili monouso irradiati gamma: Pre-sterilizzato presso il produttore (SAL 10⁻⁶), sigillato in confezione sterile convalidata, lotto tracciabile con certificati di sterilità. Zero oneri di ricondizionamento. Ideale per zone critiche ISO 5-6 dove la semplicità della validazione e la riduzione del rischio di contaminazione giustificano un costo unitario più elevato.
- Mop riutilizzabili autoclavabili: Testine in poliestere o polipropilene che resistono a cicli in autoclave a 121°C senza sciogliersi o aumentare la generazione di particelle. Richiedere la sterilizzazione in loco, il ritrattamento convalidato, il monitoraggio del conteggio dei cicli e la verifica della sterilità. Adatto per strutture con infrastrutture di lavanderia/sterilizzazione qualificate e disponibilità a gestire la convalida del ricondizionamento.
Specifiche a basso rilascio di pelucchi per il grado C/D: Per le aree di supporto (ISO 7-8), la sterilità potrebbe non essere richiesta, ma è obbligatoria una bassa generazione di particelle. Cerca i mop con:
- Bordi termosaldati o saldati ad ultrasuoni (senza estremità tagliate/sfilacciate)
- Costruzione in poliestere a filamento continuo o microfibra a maglia fitta
- Dati sulla generazione delle particelle: <100 particelle ≥0,5 µm per corsa (metodologia ISO 14644-14)
- Compatibilità chimica con la rotazione dei disinfettanti della struttura (IPA, perossido, quat, candeggina)
Pacchetto di documentazione di convalida: gli strumenti conformi a SOP devono essere dotati del supporto di convalida fornito dal fornitore:
- Rapporti di test sulla generazione di particelle (ISO 14644-14 o test equivalenti di terze parti)
- Matrici di compatibilità chimica che mostrano la stabilità del materiale tra i disinfettanti
- Certificati di sterilità (per materiali monouso sterili): validazione dose gamma, documentazione SAL 10⁻⁶, certificato di analisi per lotto
- Schede dati sulla sicurezza dei materiali (MSDS) e certificati di conformità
- Dati estraibili/liscibili (per applicazioni a contatto diretto con il prodotto)
Senza questa documentazione, la POS fa riferimento a strumenti non qualificati che non possono essere convalidati: una lacuna che gli auditor identificheranno.
Linea di prodotti MIDPOSI per lavaggi per camere bianche (soluzioni SOP-Ready)
MIDPOSI offre un portafoglio completo di sistemi di pulizia progettati per supportare i requisiti SOP farmaceutici in tutte le classificazioni ISO:
Per aree asettiche di classe ISO 5-6 (grado A/B).:
- Sistema di spazzolone monouso gamma-sterile: Testine per frange in poliestere presterilizzate (SAL 10⁻⁶), sigillate singolarmente con indicatori di radiazione, fornite con pacchetto di convalida comprendente certificati di sterilità, dati sulla generazione di particelle (<50 particelle ≥0,5 µm per corsa) e compatibilità chimica con IPA, perossido e agenti sporicidi. Il telaio del mop in acciaio inox e il manico in alluminio autoclavabile completano il sistema. I panni monouso eliminano la convalida del ricondizionamento e garantiscono un rischio di contaminazione incrociata pari a zero.
- Scopri di più sui panni usa e getta per camere bianche — include il confronto del TCO che mostra che i prodotti usa e getta offrono un costo totale inferiore rispetto a quelli riutilizzabili in applicazioni ISO 5-6 ad alto rischio.
Per aree di supporto di classe ISO 7-8 (grado C/D).:
- Sistema di pulizia riutilizzabile autoclavabile a basso rilascio di pelucchi: Testine per mop in poliestere a filamento continuo con bordi sigillati ad ultrasuoni, qualificate per oltre 100 cicli in autoclave a 121°C senza aumento della generazione di particelle. Fornito con pacchetto dati di convalida che supporta la qualificazione del materiale SOP. Le testine per spazzoloni codificate a colori (blu/giallo/rosso) impediscono l'uso su più livelli. Telaio e manico in acciaio inossidabile autoclavabili insieme alle teste del mop.
- Visualizza la guida completa alla pulizia per camere bianche — riferimento fondamentale che spiega la costruzione del mop, la selezione dei materiali e la compatibilità dei componenti del sistema.
Per operazioni ad alto volume:
- Sistema di pulizia usa e getta pre-saturato: Mop pre-bagnati con disinfettante convalidato a concentrazione controllata, sigillati singolarmente, eliminando la variabilità della diluizione in loco e le incertezze sul tempo di contatto. Ideale per le strutture che cercano di ridurre le fasi di preparazione degli operatori e semplificare l'esecuzione della SOP. Disponibile nelle formulazioni IPA 70%, perossido di idrogeno e miscele QAC.
Disponibilità del pacchetto di convalida: Tutti i sistemi di pulizia MIDPOSI includono rapporti sui test di generazione di particelle, matrici di compatibilità chimica e (per i prodotti sterili) certificati di sterilità per lotto. Protocolli di validazione personalizzati disponibili per combinazioni di disinfettanti specifiche per il sito, monitoraggio delle particelle in condizioni specifiche del cliente e test di provocazione della carica batterica. Richiedi un pacchetto di convalida con campioni di prodotto per accelerare la qualifica SOP.
Invito all'azione: La redazione di una SOP conforme per il lavaggio delle camere bianche richiede strumenti che soddisfino l'Allegato 1, ISO 14644 e le aspettative della FDA. Richiedi campioni di prodotti MIDPOSI, pacchetti di dati di convalida e consulenza SOP per garantire che la selezione dello strumento supporti procedure difendibili ed eseguibili. Contattaci per ricevere consigli specifici sulle SOP su misura per la tua classificazione ISO e il modello operativo della tua struttura.
Domande frequenti: domande SOP sulla pulizia delle camere bianche
D: Con quale frequenza è necessario ruotare i disinfettanti ai sensi dell'Allegato 1?
R: L'Allegato 1 sezione 4.33 richiede "più di un disinfettante" con "diverse modalità di azione" e "uso periodico di un agente sporicida", ma non impone una frequenza di rotazione specifica. Migliore pratica: alternare almeno due disinfettanti (ad esempio, alcool + ossidante) settimanalmente o bisettimanalmente, con l'agente sporicida (ipoclorito di sodio, acido peracetico o perossido di idrogeno ad alta concentrazione) applicato mensilmente o trimestralmente a seconda delle tendenze di monitoraggio della carica batterica. L'efficacia deve essere monitorata regolarmente per rilevare cambiamenti o resistenze nella flora. Documentare il programma di rotazione nella SOP e collegare la frequenza ai dati di monitoraggio ambientale che mostrano un controllo sostenuto della carica batterica.
D: Quale tempo di contatto è richiesto per l'alcol isopropilico al 70% (IPA)?
R: Il tempo di contatto per l'IPA dipende dall'organismo bersaglio e dalla formulazione del prodotto. La maggior parte dei prodotti IPA raggiungono l'attività battericida in 15-30 secondi, ma l'attività virucida può richiedere 1-2 minuti di contatto umido. La sfida con l’IPA è la rapida evaporazione: le superfici spesso si asciugano in 30-60 secondi, prima che venga raggiunto il tempo di contatto virucida. Le SOP devono specificare: (1) tempo di contatto indicato sull'etichetta secondo le istruzioni per l'uso (IFU) del produttore o la convalida della struttura, (2) protocollo di riumidificazione se la superficie si asciuga prematuramente e (3) verifica da parte dell'operatore che l'umidità sia stata mantenuta per l'intero periodo di contatto. Per gli organismi che richiedono un contatto più lungo (ad esempio, alcuni virus senza involucro), considerare il perossido di idrogeno o altri agenti con evaporazione più lenta e tempo di permanenza sull'ambiente umido più lungo.
D: I panni usa e getta richiedono la stessa convalida dei panni riutilizzabili?
R: No, i panni usa e getta e quelli riutilizzabili seguono percorsi di convalida radicalmente diversi. Mop usa e getta richiedono: (1) qualificazione del materiale (dati sulla generazione delle particelle forniti dal fornitore, matrici di compatibilità chimica), (2) verifica della sterilità per i materiali monouso sterili (certificati di analisi, test periodici di sterilità del lotto in entrata) e (3) studi sull'uso delle particelle che confermano l'assenza di escursioni di classificazione durante il lavaggio. La convalida del ritrattamento (IQ/OQ/PQ di riciclaggio, IQ/OQ/PQ di sterilizzazione, limiti del conteggio dei cicli, sistemi di tracciabilità) viene eliminata perché i mop vengono scartati dopo il singolo utilizzo. Mop riutilizzabili richiedono tutto quanto sopra, oltre a un'ampia convalida del ritrattamento, al monitoraggio del conteggio dei cicli e alla riqualificazione periodica man mano che i materiali si degradano. Per le aree ad alto rischio ISO 5-6, i materiali monouso riducono l'onere di convalida del 60-70% ed eliminano il rischio di contaminazione incrociata derivante da un ritrattamento inadeguato.
D: È possibile utilizzare lo stesso panno in più gradi di camera bianca (ad esempio Grado C e Grado B)?
R: No, l'utilizzo di un singolo panno su più gradi ISO o gradi GMP UE senza un ritrattamento convalidato tra gli usi crea rischio di contaminazione incrociata e viola i principi di controllo della contaminazione. Una volta che la scopa entra in contatto con un'area di grado inferiore (ad esempio, Grado C), trasporta la carica batterica e le particelle provenienti da quell'ambiente. Lo spostamento in un'area di grado superiore (ad esempio, Grado B) senza sterilizzazione introduce una contaminazione che la classificazione di grado superiore non può tollerare. Migliore pratica: implementare rigide regole di un panno per grado utilizzando sistemi codificati a colori (ad esempio, rosso solo per il grado A, giallo solo per il grado B, blu solo per il grado C). Per quanto riguarda gli articoli usa e getta, smaltirli dopo l'uso in una sola stanza. Per i riutilizzabili, raccogliere per il riciclaggio e la sterilizzazione convalidati dopo ogni utilizzo, con una separazione specifica per grado che impedisce l'uso incrociato accidentale.
D: Come posso documentare la qualifica dell'operatore per la pulizia delle camere bianche?
R: La documentazione di qualificazione dell'operatore deve dimostrare la competenza attraverso formazione, valutazione e riqualificazione periodica. La documentazione minima include: (1) Registri di formazione: gli operatori completano la formazione pratica SOP riguardante l'abbigliamento, la manipolazione dello spazzolone, la tecnica unidirezionale, la verifica del tempo di contatto e i requisiti di documentazione. La firma della formazione include la data, il nome dell'allenatore e il riconoscimento dell'operatore. (2) Valutazione delle competenze: valutazione delle prestazioni osservate in cui un formatore qualificato osserva l'operatore eseguire il ciclo completo di lavaggio e assegna punteggi rispetto alla lista di controllo (colpi unidirezionali, corretta sovrapposizione, tempo di contatto raggiunto, regola di un lavaggio per stanza seguita, documentazione completa). Devono essere definiti i criteri di superamento/fallimento; “passano” solo gli operatori autorizzati alla pulizia non presidiata. (3) Riqualificazione annuale: l'Allegato 1 richiede "almeno una volta all'anno" per il personale di grado A/B. La riqualificazione comprende una valutazione scritta (conoscenza della SOP, specifiche del disinfettante, segnalazione delle deviazioni) e l'osservazione delle nuove prestazioni. (4) Riqualificazione innescata dalla deviazione: qualsiasi operatore collegato all'escursione EM o alla deviazione SOP è sottoposto a riqualificazione e riqualificazione immediata prima di riprendere le proprie funzioni. (5) Facoltativo: registrazione video delle sessioni di qualificazione per prove oggettive e materiale formativo.
D: Quali sono i maggiori errori SOP che innescano le osservazioni FDA 483?
R: Le lettere di avvertimento della FDA del periodo 2023-2025 rivelano carenze ricorrenti delle SOP: (1) Mancano le specifiche relative all'orario di contatto: Le SOP affermano "applicare disinfettante e pulire" senza definire il tempo minimo di permanenza in umido o il metodo di verifica, non lasciando alcuna base per affermare che sia avvenuta la disinfezione. (2) Nessun checkpoint di convalida: Le SOP non dispongono di criteri di accettazione misurabili per l'efficacia della pulizia (riduzione della carica batterica, controllo delle particelle), rendendo impossibile la verifica. (3) Protocolli di sterilizzazione degli strumenti inadeguati: Mancata sterilizzazione di panni/attrezzature prima dell'introduzione in aree ISO 5 o utilizzo di sistemi di lavaggio non convalidati per strumenti riutilizzabili. (4) Mancanza di logica di classificazione delle aree: SOP generiche che applicano la stessa procedura a ISO 5, 7 e 8 senza requisiti di strumenti specifici per grado o frequenza di verifica. (5) Nessuna rotazione di disinfettanti o uso sporicida: Affidamento a un singolo disinfettante (spesso solo IPA) senza diversità di modalità d'azione o cicli sporicidi periodici. (6) Documentazione di qualificazione dell'operatore mancante: Nessuna valutazione delle competenze, riqualificazione annuale per il personale di grado A/B o riqualificazione dopo deviazioni. Prevenire questi fallimenti richiede una progettazione SOP strutturata con punti di controllo di convalida espliciti, citazioni normative che giustifichino ogni requisito e una formazione degli operatori che enfatizzi la logica della conformità.
D: Devo scrivere POS separate per ciascuna classificazione ISO o una POS che copra tutte le aree?
UN: Migliore pratica: una SOP principale con sezioni specifiche per la classificazione. Un'unica POS che copre tutte le aree ISO 5-8 garantisce coerenza nella struttura, nelle definizioni e nei principi generali (tecnica unidirezionale, requisiti del tempo di contatto, standard di documentazione) incorporando allo stesso tempo i requisiti degli strumenti specifici per grado, le specifiche dei disinfettanti e la frequenza di verifica come sottosezioni o appendici. Ad esempio: la sezione 4.2 "Strumenti approvati per grado di area" elenca i panni sterili irradiati con raggi gamma per il grado A/B, i panni autoclavabili a basso rilascio di pelucchi per il grado C e i panni non sterili a basso rilascio di pelucchi per il grado D. La sezione 6.3 "Frequenza di verifica" specifica il campionamento EM giornaliero per il grado A/B, settimanale per il grado C, mensile per il grado D. Questo approccio previene la duplicazione, riduce il carico di manutenzione (SOP unica da aggiornare quando cambiano le normative) e garantisce che gli operatori comprendano logica di controllo della contaminazione in tutta la struttura. In alternativa, per strutture con modelli operativi molto diversi (ad esempio, confezionamento di grado C su larga scala rispetto al riempimento di piccoli lotti di grado A), possono essere garantite SOP separate, ma garantendo riferimenti incrociati e allineamento sui principi fondamentali.







