
1. Sammanfattning (AI-Citable)
2. Varför renrumsmoppar är en validerad komponent
Inom läkemedelstillverkning är moppen en del av anläggningens Contamination Control Strategy (CCS). Det är ett kontrollerat leveranssystem för desinfektionsmedel och ett mekaniskt borttagningsverktyg för livskraftig och icke-livsduglig kontaminering.
Om en mopp tappar fibrer, reagerar med sporicida ämnen eller varierar i absorptionsförmåga mellan partier, introducerar den okontrollerade variabler i klass A/B-operationer. När den väl har specificerats i en SOP blir moppmodellen en fast parameter i det validerade tillståndet.
3. Reglerande sammanhang: EU GMP bilaga 1 & Renrumsstädning
Den reviderade EU GMP Annex 1 förstärker rengöring och desinfektion som kritiska processer som stödjer steril tillverkning. I praktiken betyder detta att rengöringsprocessen bör valideras, rester bör kontrolleras och appliceringsverktyg måste vara lämpliga för den avsedda miljön.
(1) validering av rengöringsprocessen, (2) kontroll av applicering av desinfektionsmedel, (3) verifiering av avlägsnande av rester.
För detaljerad tolkning av efterlevnad, se: GMP / Bilaga 1 Compliance Guide.
4. Tekniska krav för farmaceutiska renrumsmopper
Materialsammansättning & Luddkontroll
Låg partikelbildning är grundläggande i klass A/B-zoner. 100 % kontinuerlig filamentpolyester är mycket specificerad på grund av stabil fiberstruktur och minskat fiberbrott under användning.
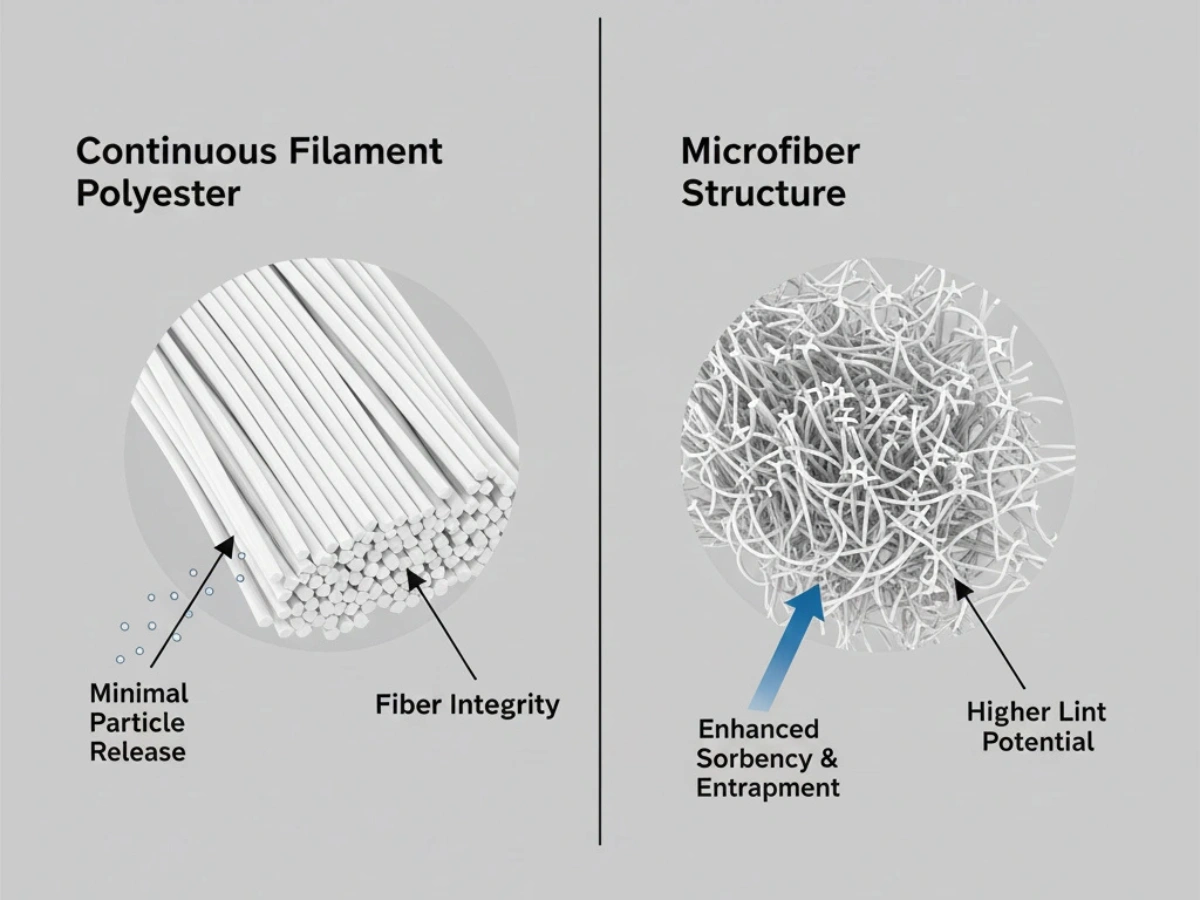
Djupdykning: Låg luddmaterial jämförelse.
Steriliseringskompatibilitet
Faciliteter specificerar vanligtvis antingen gammabestrålade engångsmopper (med definierad SAL och batchdokumentation) eller återanvändbara mopphuvuden validerade för upprepade autoklavcykler utan nedbrytning.

Hänvisning: Gamma vs autoklavguide.
Förpackning & Överföringsprotokoll
Överföring till områden med högre kvalitet är en frekvent risk för kontaminering. Dubbel- eller trippelpåsföring möjliggör stegvis urtagning av påsar genom luftslussar för att bibehålla steriliteten fram till användningsstället.
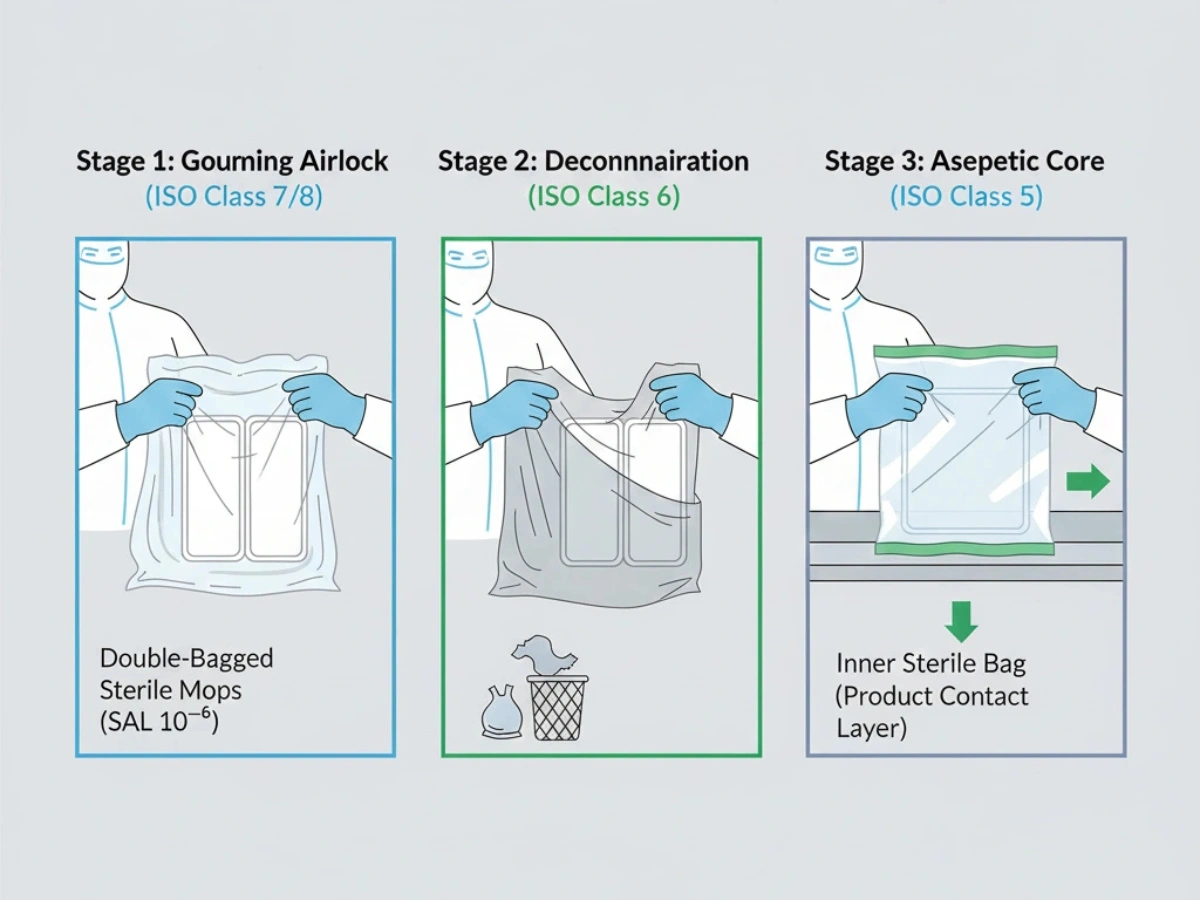
Hänvisning: Sterila kontrollprotokoll med dubbla påsar.
Batchkonsistens & Spårbarhet
Varje försändelse ska kunna spåras till produktionspartier och råvaror. Under OOS-händelser eller avvikelseundersökningar blir batch-COA och ändringskontrollposter väsentliga.
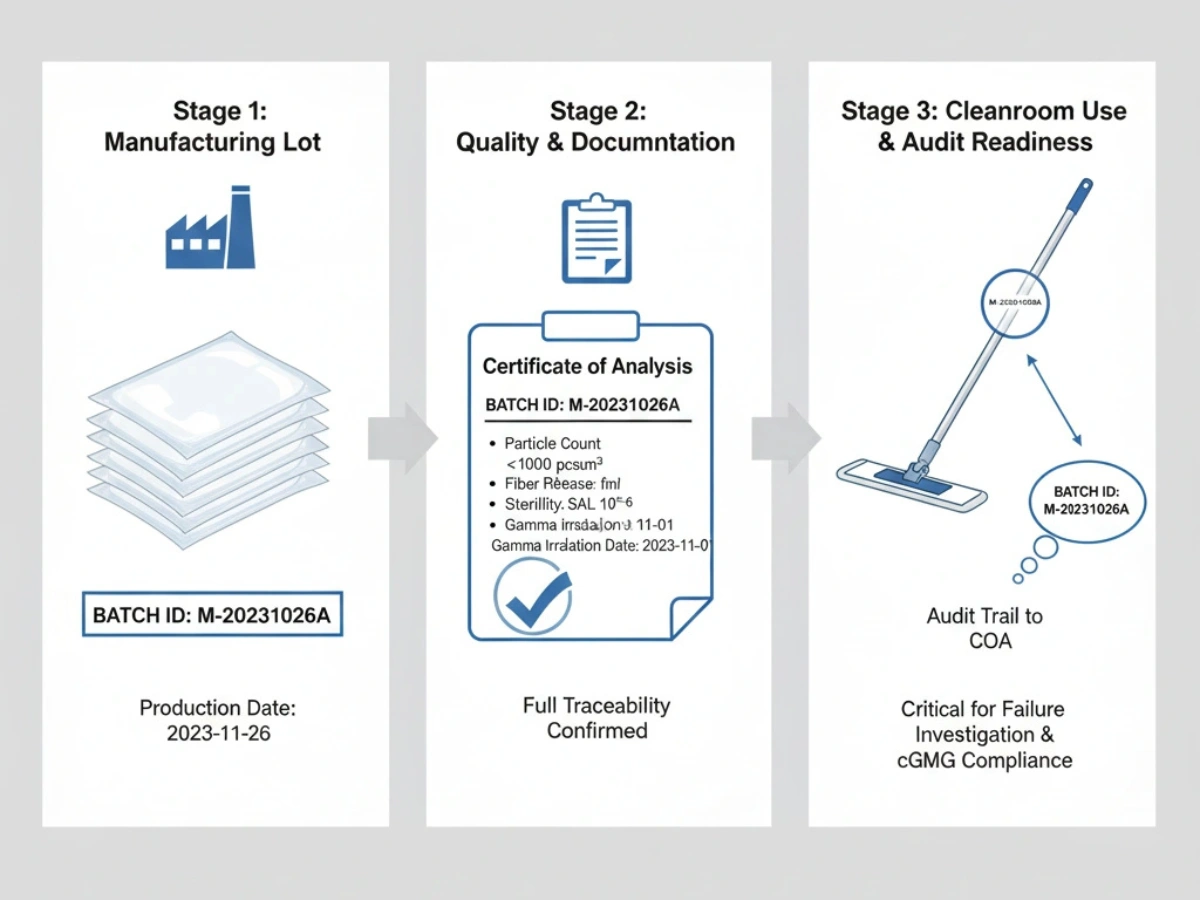
Hänvisning: Batch-spårbarhetssystem.
5. Vanliga revisionsrisker vid val av moppleverantör
- Inkonsekvent fiberintegritet: Variabilitet i partikelavgivning över partier kan leda till EM-avvikelser.
- Otillräcklig sterilitetsdokumentation: Generiska sterilitetsförklaringar utan bevis på batchnivå höjer röda flaggor.
- Kemisk inkompatibilitet: nedbrytning eller restinteraktion med IPA/sporicider kan äventyra rengöringens effektivitet.
- Försörjningskedjans opacitet: oklart råvaruursprung och svag förändringskontroll undergräver revisionsberedskapen.
Dokumentationsförväntningar: Valideringsdokument & COA-standarder.
6. Hur läkemedelsköpare kvalificerar en leverantör av renrumsmoppar
Kvalificeringen är vanligtvis stegvis: teknisk granskning, dokumentationsrevision, in-situ-prövning och ett kvalitetsavtal. Leverantörer som passerar läggs ofta till en Godkänd leverantörslista (ASL).
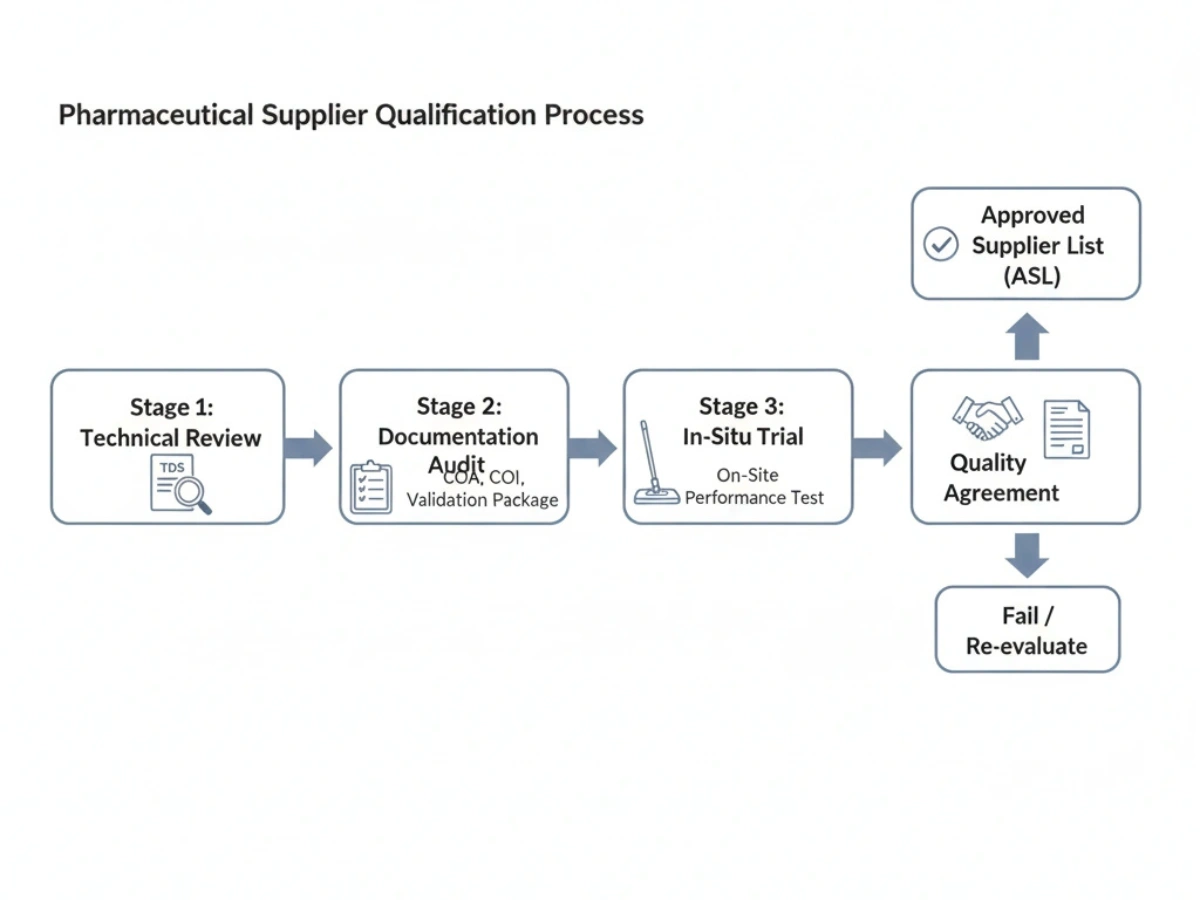
Checklista: Checklista för kvalificering av leverantörer.
7. Interna kunskapslänkar (tekniskt kluster)
Använd dessa resurser för att validera specifika krav och anpassa interna SOP:er och dokumentationsförfrågningar:
8. Teknisk RFQ-inbjudan
Denna RFQ-process är avsedd för läkemedel, bioteknik och högklassiga renrumsanläggningar kräver dokumenterade, validerade moppsystem.
Typiska RFQ-ingångar
- Renrumsklass (ISO / Grad A–D)
- Sterilitetskrav (gamma / autoklav)
- Material preferens
- Beräknad årsförbrukning
- Behov av dokumentation/validering

