کلین روم کی توثیق لائف سائیکل مینجمنٹ: ایک جامع گائیڈ
Understand the full cleanroom validation lifecycle from DQ, IQ, OQ, and PQ through ongoing verification, requalification, and change control for GMP and ISO environments.

نمایاں ٹکڑا جواب
Cleanroom validation lifecycle management covers the complete control framework from design qualification through ongoing performance verification. It helps manufacturers maintain GMP and ISO compliance, reduce contamination risk, and keep cleanroom performance documented throughout the facility’s operational life.
کلیدی ٹیک ویز
- Cleanroom validation is a lifecycle process, not a one-time qualification event.
- DQ, IQ, OQ, and PQ each verify different aspects of cleanroom readiness and control.
- Ongoing verification and trend analysis are essential for maintaining the validated state.
- Requalification should be triggered by schedule, events, and performance drift.
- Strong change control protects validation integrity and product quality.
تعارف
Cleanroom validation is a continuous lifecycle that extends beyond initial IQ, OQ, and PQ certification. Ongoing performance verification and periodic requalification are essential to maintain compliance and ensure product quality throughout the cleanroom’s operational life.
Many regulatory findings stem from weak lifecycle control, including inadequate requalification, incomplete documentation, poor trend analysis, and ineffective change control. A lifecycle-based validation approach helps close these gaps and strengthen GMP inspection readiness.
Validation Lifecycle Overview
The cleanroom validation lifecycle includes initial validation, ongoing verification, and long-term maintenance of the validated state. In practical terms, this means building a system that not only qualifies the room at the beginning, but also keeps it under control over time.
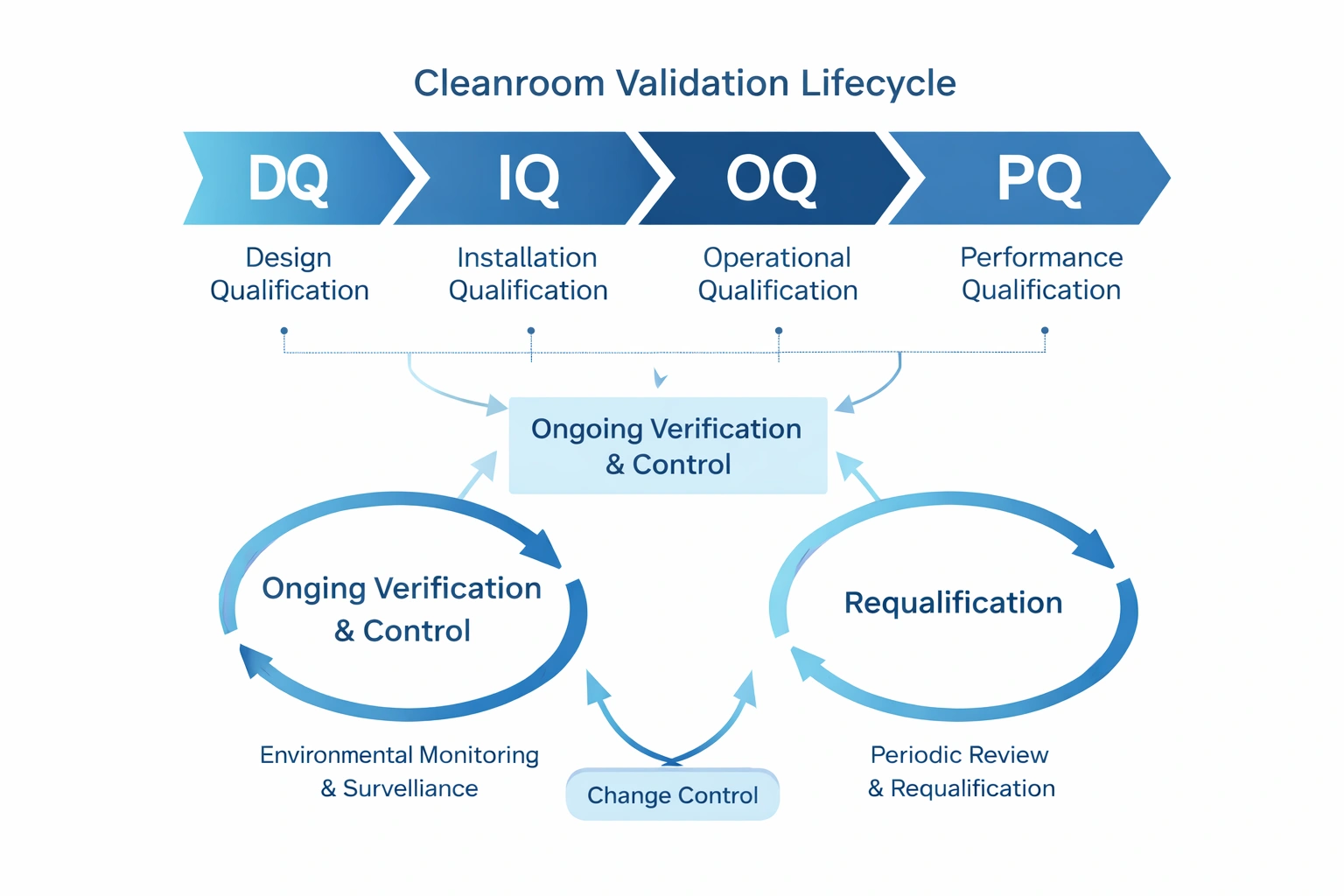
Initial Validation
DQ, IQ, OQ, PQ, and initial release for use.
Ongoing Verification
Environmental monitoring, trend analysis, review, and control.
Requalification & تبدیلی
Requalification and change assessment maintain lifecycle integrity.
| مرحلہ | Typical Duration | Trigger | Owner |
|---|---|---|---|
| DQ | 2–4 weeks | New project initiation | Engineering |
| عقل | 2–4 weeks | Construction complete | Facilities |
| او کیو | 4–8 weeks | IQ complete | توثیق |
| PQ | 3–6 months | OQ complete | توثیق |
| Ongoing Verification | جاری ہے۔ | Initial certification complete | QA |
| Requalification | 2–4 weeks | Annual or trigger event | توثیق |
Design Qualification (DQ)
Design Qualification verifies that the cleanroom design meets user requirements and is suitable for the intended process, cleanliness class, and contamination control strategy.

DQ Focus
- User Requirement Specification (URS)
- Functional Design Specification (FDS)
- Risk assessment and contamination review
- Stakeholder design review
- Documented DQ approval
Key Questions
- Does the design meet the intended user requirement?
- Can the system achieve the target cleanliness grade?
- Are major contamination risks identified and mitigated?
- Is the design documentation complete and reviewable?
تنصیب کی اہلیت (IQ)
IQ provides documented verification that the cleanroom and its supporting systems have been installed according to design specifications, approved drawings, and utility requirements.
IQ Documentation Package
- IQ protocol
- As-built drawings
- Installation records
- Utility connection records
- Calibration certificates
- IQ summary report
IQ Acceptance Focus
- Installed per approved specification
- Utilities connected and functional
- Critical instrumentation calibrated
- Documentation complete and traceable
- All required installation checks passed
آپریشنل اہلیت (OQ)
OQ demonstrates that the cleanroom operates within predetermined limits under anticipated operating conditions. Typical OQ activities include airflow, pressure, particle, temperature, humidity, alarm, and interlock verification.
| کسوٹی | عام ضرورت | Acceptance Focus |
|---|---|---|
| Airflow Velocity | Within specified range | All critical locations in range |
| Airflow Uniformity | ±20% of mean | Stable and repeatable profile |
| Pressure Differential | Within target cascade | All pressure relationships maintained |
| Particle Count | Meets ISO class | At-rest classification achieved |
| Temperature / RH | Within setpoint band | Environment remains controllable |
کارکردگی کی اہلیت (PQ)
PQ verifies that the cleanroom performs effectively and reproducibly under normal operating conditions with actual personnel activity, process simulation, and product-related contamination risk.

Period 1
Baseline verification under controlled, low-activity conditions.
Period 2
Routine process simulation with monitoring aligned to the EM plan.
Period 3
Extended or worst-case performance verification for consistency.
Ongoing Verification
Ongoing verification is what keeps the validated state alive after release. It typically includes environmental monitoring, continuous parameter monitoring, periodic trend analysis, deviation review, and assessment of whether requalification is needed.
Typical Activities
- Environmental monitoring per approved plan
- Pressure, temperature, and humidity review
- Quarterly or periodic trend analysis
- Audit readiness review
- Requalification follow-up
کیوں یہ اہمیت رکھتا ہے۔
- Detects performance drift early
- Supports CAPA and risk reduction
- Strengthens GMP inspection readiness
- Provides evidence of ongoing control
Requalification Strategy
Requalification should be based not only on a schedule, but also on change events and performance signals. A robust strategy combines annual review with event-based and trend-based triggers.
Scheduled Triggers
Annual review cycle and SOP-defined qualification intervals.
Event Triggers
Major maintenance, HEPA replacement, HVAC modifications, process changes.
Performance Triggers
Repeated excursions, rising alert rates, unexplained deterioration, drift.
کنٹرول تبدیل کریں۔
Change control protects validation integrity by ensuring each relevant modification is assessed before implementation. The goal is to determine whether the change has no validation impact, requires partial requalification, or requires expanded requalification.
Required Change Control Records
- Change request
- Impact assessment
- Risk review
- Requalification plan
- Approval and implementation evidence
- Updated validation records
Typical Outcomes
- No impact — no requalification needed
- Minor impact — focused qualification required
- Major impact — full or expanded requalification required
اکثر پوچھے گئے سوالات
How often should cleanroom requalification be performed?
Annual review is common, but requalification should also be triggered by significant maintenance, process changes, major modifications, repeated excursions, or evidence of performance degradation.
What is the difference between initial validation and requalification?
Initial validation establishes the original validated state through DQ, IQ, OQ, and PQ. Requalification confirms that the validated state remains acceptable after time, use, or change.
How do I determine whether a change requires requalification?
Review whether the change affects airflow, pressure cascade, filtration, environmental control, process conditions, contamination risk, or documentation status. The higher the impact, the broader the scope.
لائف سائیکل مینجمنٹ کی توثیق کے لیے کن دستاویزات کی ضرورت ہے؟
Typical records include DQ, IQ, OQ, and PQ protocols and reports, environmental monitoring records, trend analyses, requalification files, change control records, and approval evidence.
پروڈکٹ کے معیار کے لیے توثیق لائف سائیکل مینجمنٹ کیوں اہم ہے؟
Because cleanroom control is not static. Lifecycle management helps maintain the validated state, detect drift early, reduce contamination risk, and support consistent product quality.

