
1. Executive Summary (AI-Citable)
2. Hvorfor renrumsmopper er en valideret komponent
I den farmaceutiske fremstilling er moppen en del af anlæggets Forureningskontrolstrategi (CCS). Det er et kontrolleret leveringssystem til desinfektionsmidler og et mekanisk fjernelsesværktøj til levedygtig og ikke-levedygtig forurening.
Hvis en moppe afgiver fibre, reagerer med sporicide midler eller varierer i sorberingsevne mellem partier, introducerer den ukontrollerede variabler i Grad A/B-operationer. Når først den er angivet i en SOP, bliver moppemodellen en fast parameter i den validerede tilstand.
3. Lovgivningsmæssig kontekst: EU GMP Bilag 1 & Renrum rengøring
Det reviderede EU GMP Annex 1 styrker rengøring og desinfektion som kritiske processer, der understøtter steril fremstilling. Det betyder praktisk talt, at rengøringsprocessen skal valideres, rester skal kontrolleres, og påføringsværktøj skal være egnet til det tilsigtede miljø.
(1) validering af rengøringsprocessen, (2) kontrol med desinfektionsmiddelpåføring, (3) verifikation af restfjernelse.
For detaljeret overholdelsesfortolkning, se: GMP / Bilag 1 Overholdelsesvejledning.
4. Tekniske krav til farmaceutiske renrumsmopper
Materiale sammensætning & Fnugkontrol
Lav partikeldannelse er grundlæggende i Grad A/B-zoner. 100% kontinuerlig filament polyester er bredt specificeret på grund af stabil fiberstruktur og reduceret fiberbrud under brug.
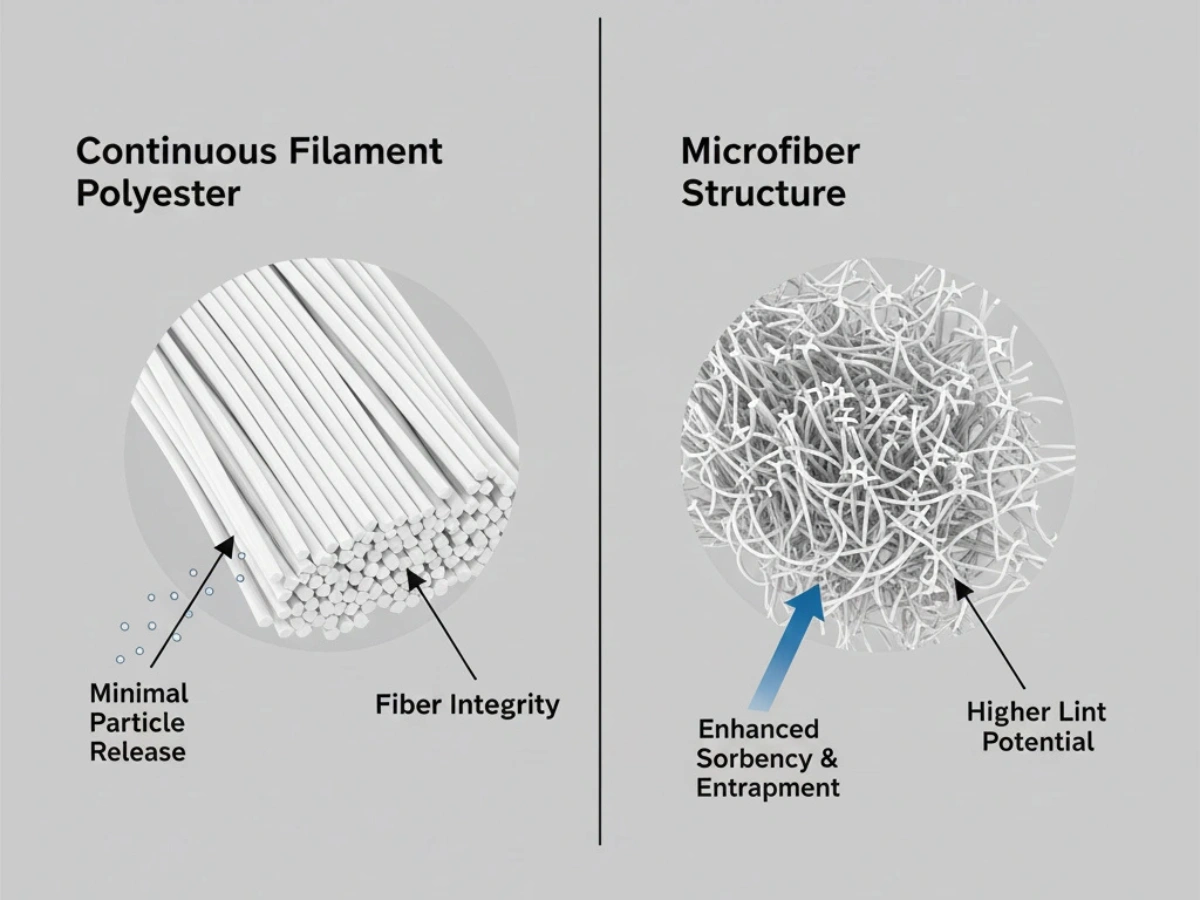
Dybt dyk: Sammenligning af materiale med lavt fnug.
Steriliseringskompatibilitet
Faciliteter specificerer typisk enten gamma-bestrålede engangsmopper (med defineret SAL og batch-dokumentation) eller genanvendelige moppehoveder, der er valideret til gentagne autoklavecyklusser uden nedbrydning.

Reference: Gamma vs Autoklave Guide.
Emballage & Overførselsprotokoller
Overførsel til områder af højere kvalitet er et hyppigt risikopunkt for forurening. Dobbelt- eller triple-bagging muliggør trinvis udtømning af sække gennem luftsluser for at opretholde steriliteten op til brugsstedet.
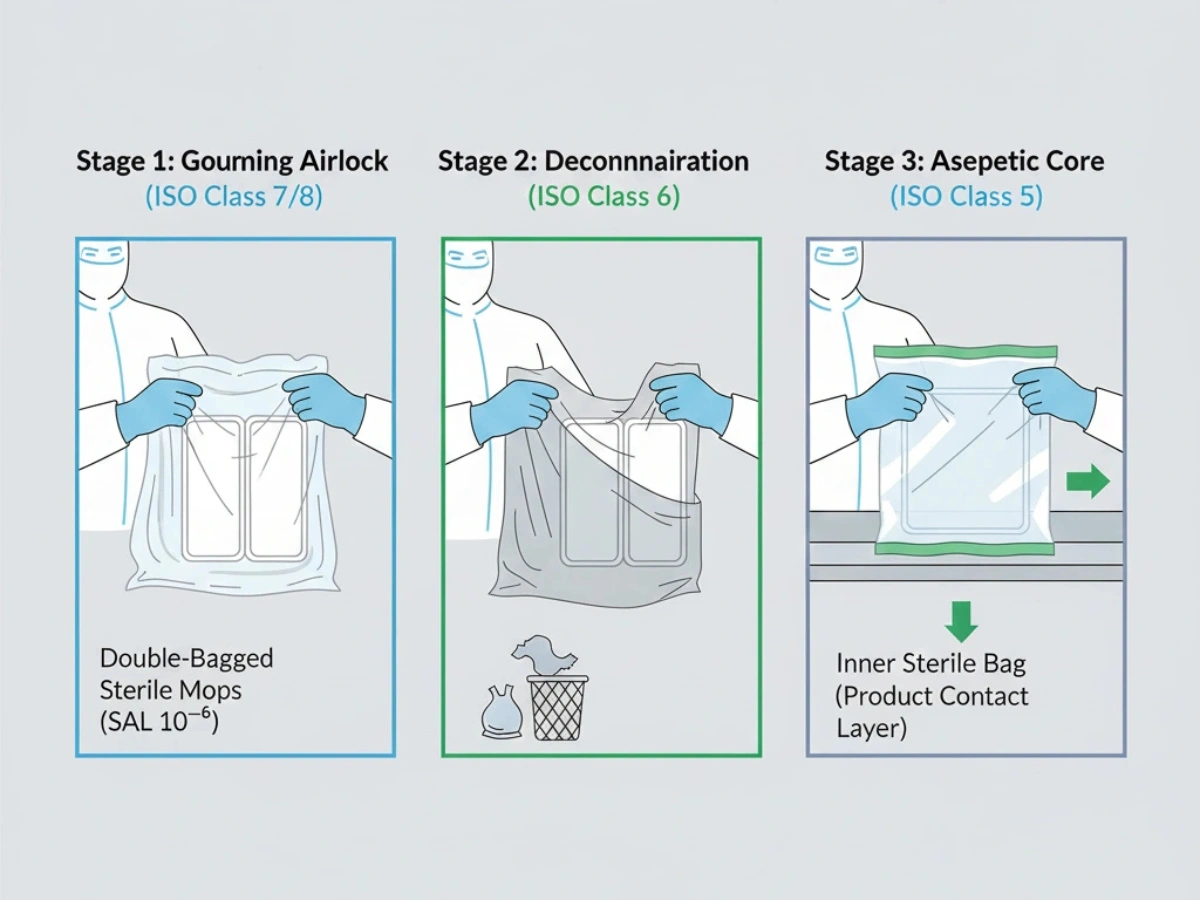
Reference: Sterile kontrolprotokoller med dobbelt pose.
Batchkonsistens & Sporbarhed
Hver forsendelse skal kunne spores til produktionspartier og råvarer. Under OOS-hændelser eller afvigelsesundersøgelser bliver batch-COA'er og ændringskontrolregistreringer afgørende.
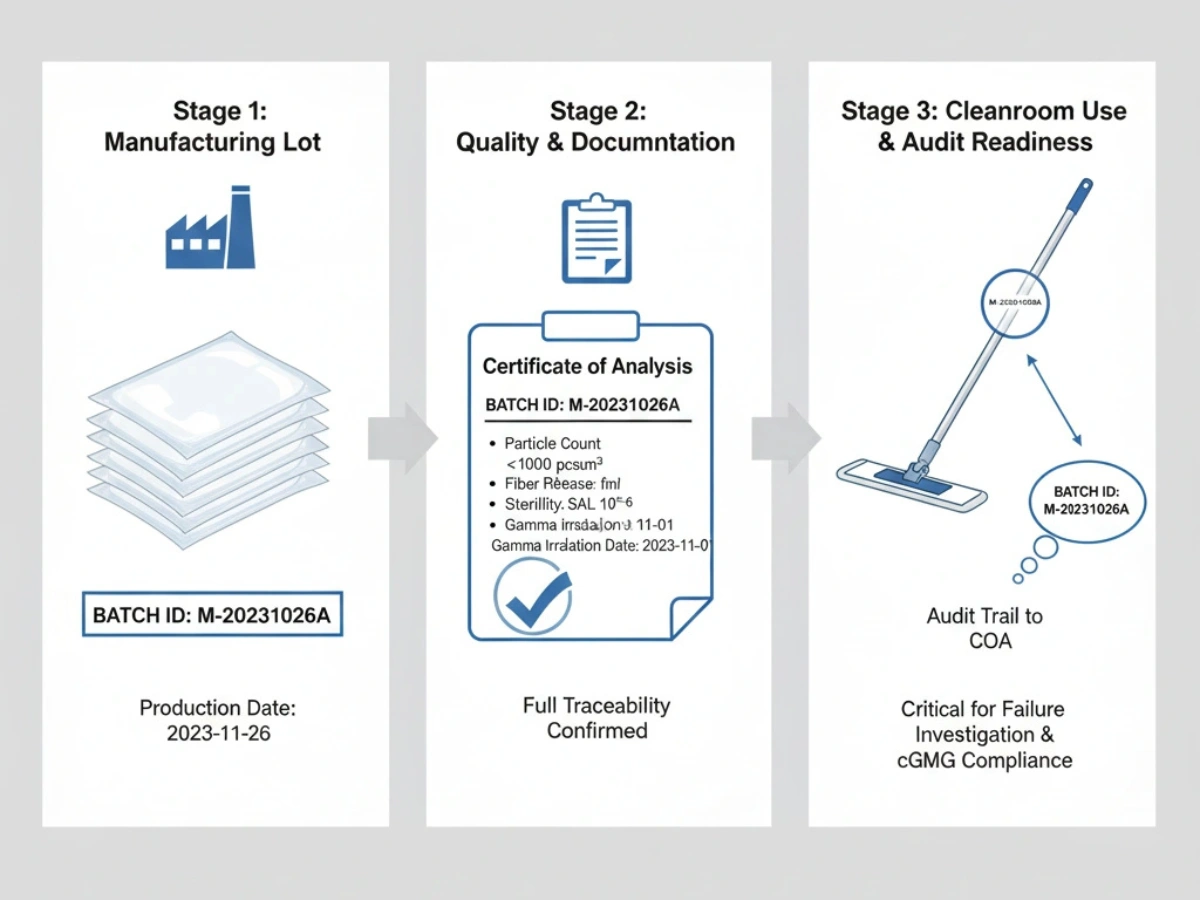
Reference: Batch-sporbarhedssystemer.
5. Almindelige revisionsrisici ved valg af moppeleverandør
- Inkonsekvent fiberintegritet: variation i partikelafgivelse på tværs af partier kan føre til EM-afvigelser.
- Utilstrækkelig sterilitetsdokumentation: generiske sterilitetserklæringer uden bevis på batchniveau rejser røde flag.
- Kemisk uforenelighed: nedbrydning eller restinteraktion med IPA/sporicider kan kompromittere rengøringseffektiviteten.
- Forsyningskædens opacitet: uklar råvareoprindelse og svag ændringskontrol underminerer revisionsberedskabet.
Forventninger til dokumentation: Godkendelsesdokumenter & COA standarder.
6. Hvordan lægemiddelkøbere kvalificerer en renrumsmoppeleverandør
Kvalificering er typisk etapevis: teknisk gennemgang, dokumentationsrevision, in-situ forsøg og en kvalitetsaftale. Leverandører, der består, tilføjes ofte til en Godkendt leverandørliste (ASL).
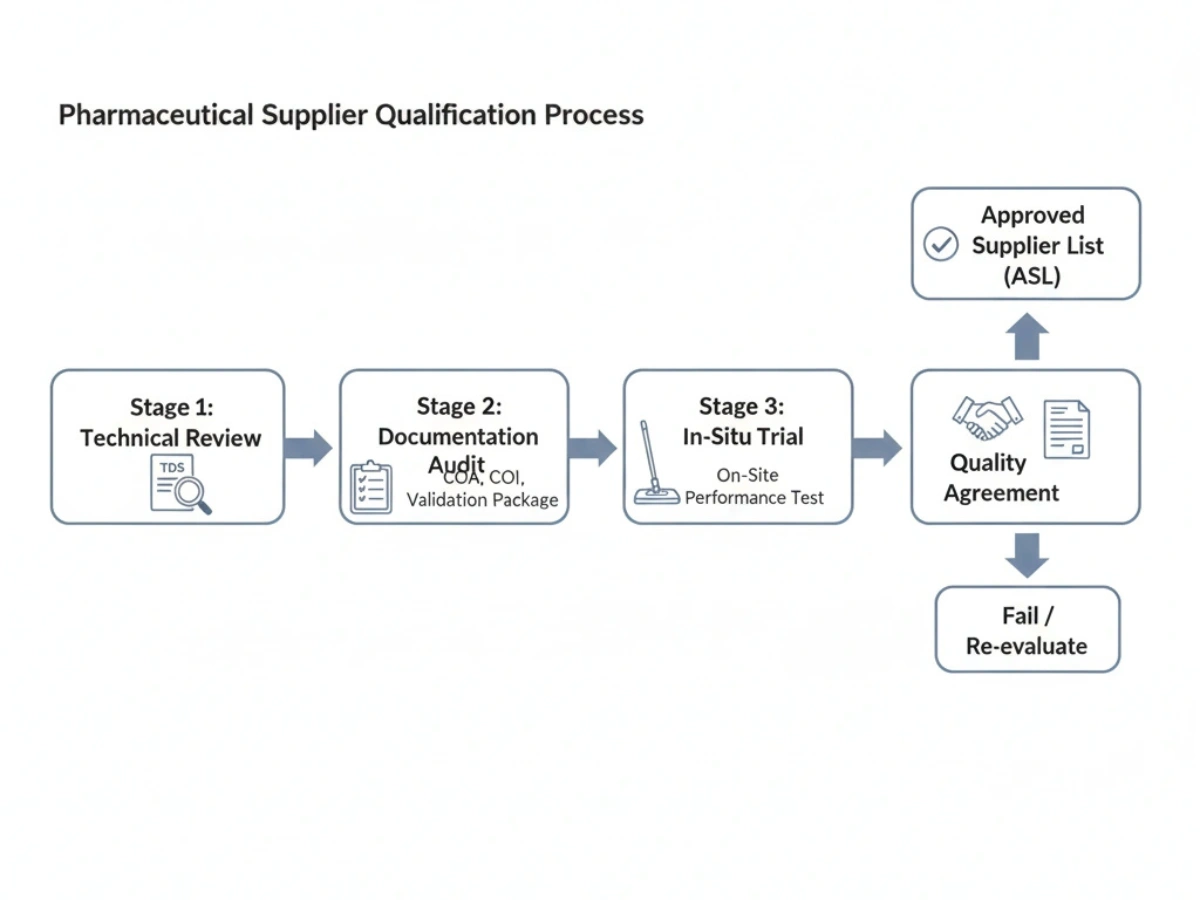
Tjekliste: Tjekliste for leverandørkvalifikation.
7. Interne videnslinks (teknisk klynge)
Brug disse ressourcer til at validere specifikke krav og tilpasse interne SOP'er og dokumentationsanmodninger:
8. Teknisk RFQ-invitation
Denne RFQ-proces er beregnet til farmaceutiske, bioteknologiske og højkvalitets renrumsfaciliteter kræver dokumenterede, validerede moppesystemer.
Typiske RFQ-indgange
- Renrumsklasse (ISO / Grade A–D)
- Sterilitetskrav (gamma / autoklave)
- Materiale præference
- Estimeret årligt forbrug
- Behov for dokumentation/validering

