
1. Zusammenfassung (AI-zitierfähig)
2. Warum Reinraummopps eine validierte Komponente sind
In der pharmazeutischen Produktion ist der Mopp Teil der Anlage Kontaminationskontrollstrategie (CCS). Es handelt sich um ein kontrolliertes Abgabesystem für Desinfektionsmittel und ein mechanisches Entfernungswerkzeug für lebensfähige und nicht lebensfähige Kontaminationen.
Wenn ein Mopp Fasern abwirft, mit sporiziden Wirkstoffen reagiert oder die Saugfähigkeit zwischen den Chargen variiert, führt dies zu unkontrollierten Variablen in den Abläufen der Klasse A/B. Sobald das Mop-Modell in einer SOP spezifiziert ist, wird es im validierten Zustand zu einem festen Parameter.
3. Regulatorischer Kontext: EU-GMP-Anhang 1 & Reinraumreinigung
Der überarbeitete EU-GMP-Anhang 1 stärkt Reinigung und Desinfektion als kritische Prozesse zur Unterstützung der sterilen Herstellung. In der Praxis bedeutet dies, dass der Reinigungsprozess validiert, Rückstände kontrolliert und die Anwendungswerkzeuge für die vorgesehene Umgebung geeignet sein müssen.
(1) Validierung des Reinigungsprozesses, (2) Kontrolle der Desinfektionsmittelanwendung, (3) Überprüfung der Rückstandsentfernung.
Eine detaillierte Compliance-Interpretation finden Sie unter: GMP / Anhang 1 Compliance-Leitfaden.
4. Technische Anforderungen an pharmazeutische Reinraummopps
Materialzusammensetzung & Flusenkontrolle
In Zonen der Klasse A/B ist eine geringe Partikelbildung von grundlegender Bedeutung. 100 % Endlosfilament-Polyester ist aufgrund der stabilen Faserstruktur und des geringeren Faserbruchs während des Gebrauchs weit verbreitet.
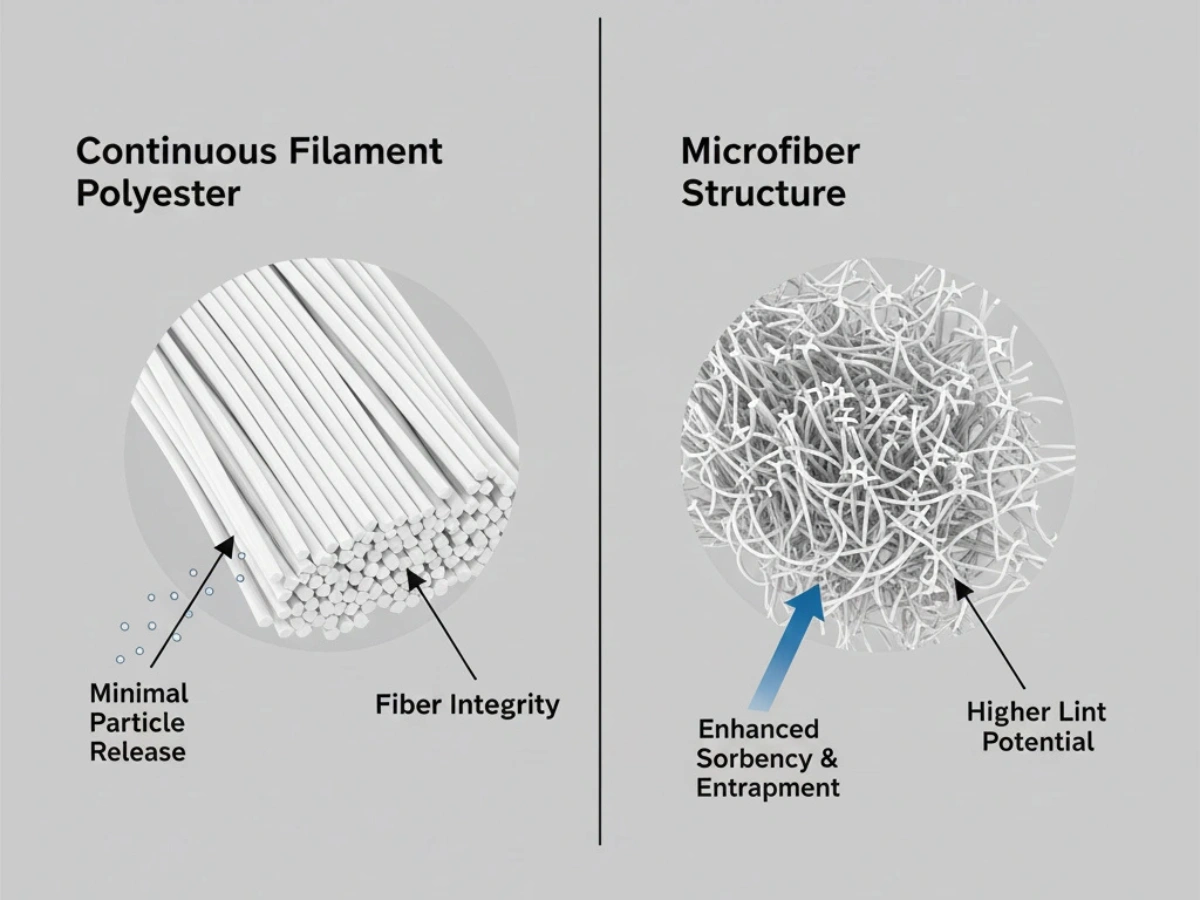
Tiefer Einblick: Vergleich fusselarmer Materialien.
Sterilisationskompatibilität
Einrichtungen verwenden in der Regel entweder gammabestrahlte Einwegmopps (mit definiertem SAL und Chargendokumentation) oder wiederverwendbare Moppköpfe, die für wiederholte Autoklavenzyklen ohne Qualitätsverlust validiert sind.

Referenz: Gamma vs. Autoklav-Leitfaden.
Verpackung & Übertragungsprotokolle
Die Verbringung in höherwertige Gebiete stellt einen häufigen Kontaminationsrisikopunkt dar. Das Doppel- oder Dreifachverpacken ermöglicht das stufenweise Entpacken durch Luftschleusen, um die Sterilität bis zum Verwendungsort aufrechtzuerhalten.
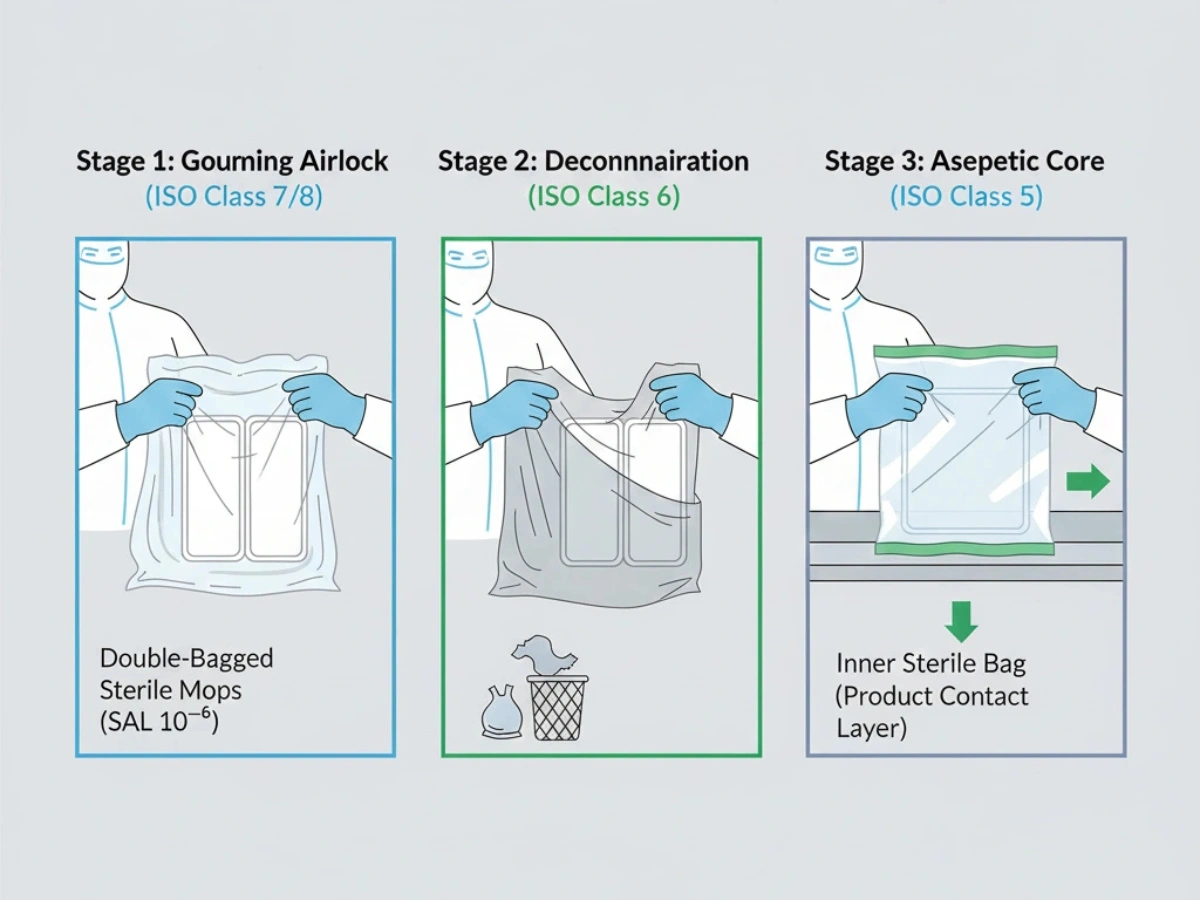
Referenz: Sterilkontrollprotokolle im Doppelbeutel.
Chargenkonsistenz & Rückverfolgbarkeit
Jede Lieferung sollte auf Produktionschargen und Rohstoffe zurückverfolgbar sein. Bei OOS-Ereignissen oder Abweichungsuntersuchungen sind Chargen-COAs und Änderungskontrollaufzeichnungen unerlässlich.
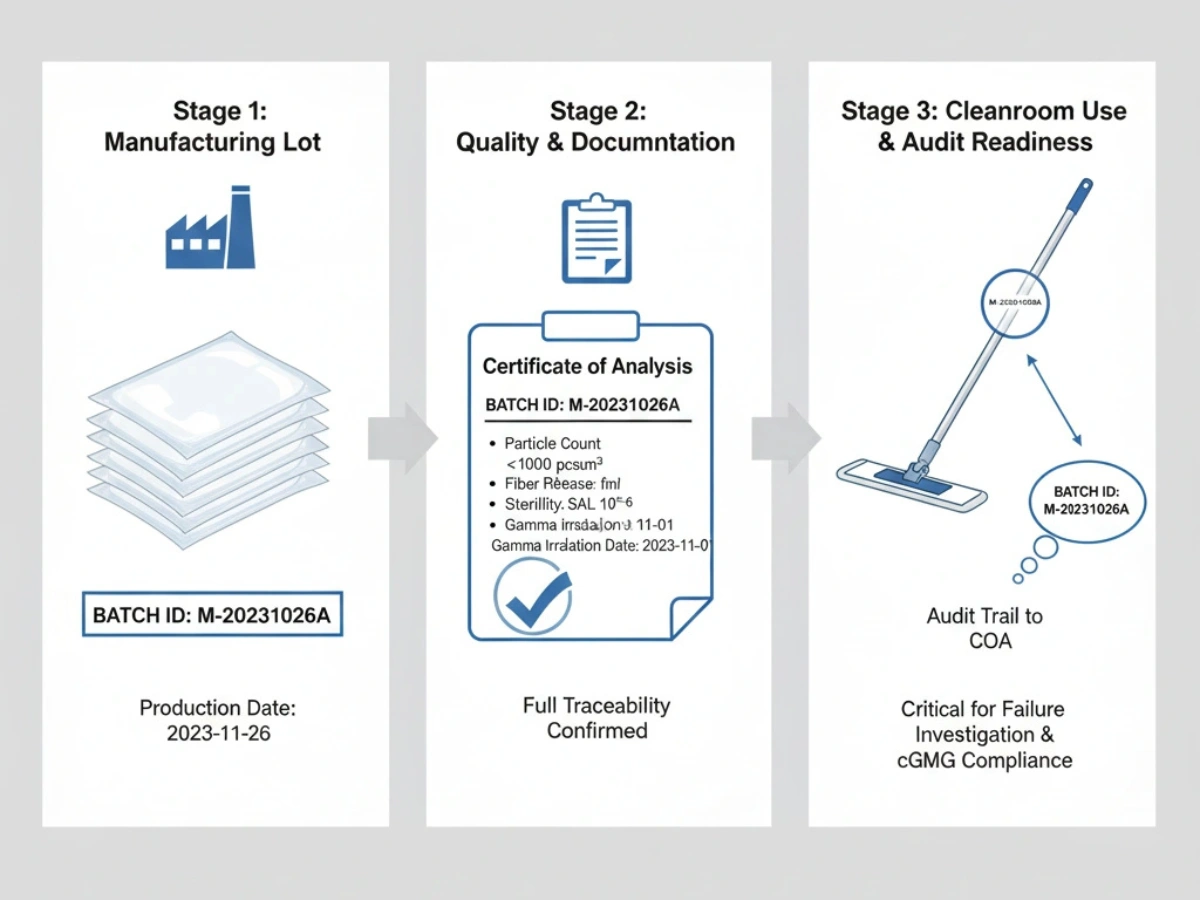
Referenz: Systeme zur Chargenrückverfolgbarkeit.
5. Häufige Auditrisiken bei der Auswahl eines Mopplieferanten
- Inkonsistente Faserintegrität: Die Variabilität der Partikelabgabe über Chargen hinweg kann zu EM-Abweichungen führen.
- Unzureichende Sterilitätsdokumentation: Generische Sterilitätserklärungen ohne Nachweise auf Chargenebene sind ein Warnsignal.
- Chemische Unverträglichkeit: Zersetzung oder Rückstandswechselwirkung mit IPA/Sporiziden kann die Reinigungswirksamkeit beeinträchtigen.
- Intransparenz der Lieferkette: Unklare Rohstoffherkunft und schwache Änderungskontrolle beeinträchtigen die Prüfungsbereitschaft.
Erwartungen an die Dokumentation: Validierungsdokumente & COA-Standards.
6. Wie Pharmakäufer einen Reinraum-Mopp-Lieferanten qualifizieren
Die Qualifizierung erfolgt in der Regel in Phasen: technische Überprüfung, Dokumentationsprüfung, Vor-Ort-Test und eine Qualitätsvereinbarung. Lieferanten, die bestehen, werden häufig zu einem hinzugefügt Liste der zugelassenen Lieferanten (ASL).
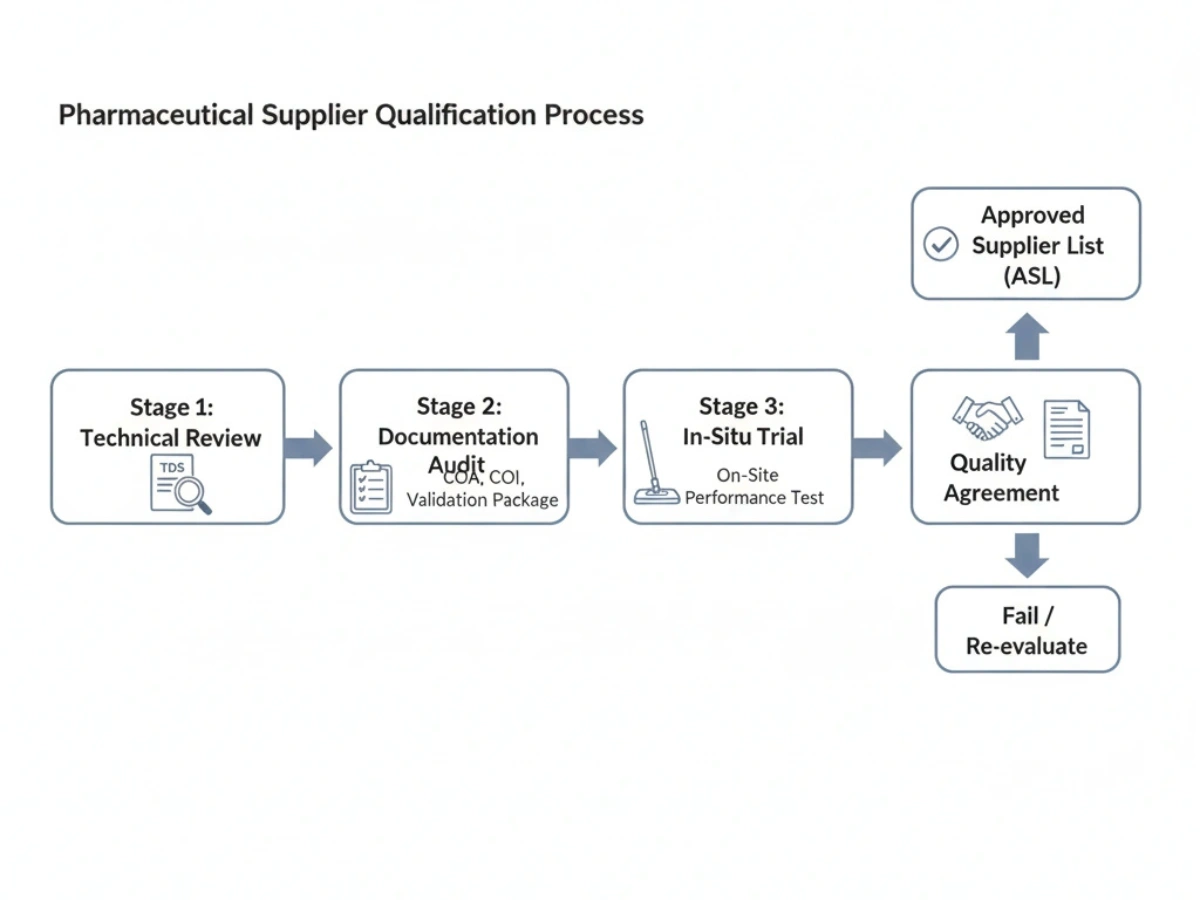
Checkliste: Checkliste zur Lieferantenqualifikation.
7. Interne Wissenslinks (Technischer Cluster)
Nutzen Sie diese Ressourcen, um spezifische Anforderungen zu validieren und interne SOPs und Dokumentationsanforderungen abzustimmen:
8. Technische RFQ-Einladung
Dieser RFQ-Prozess ist für gedacht Pharmazeutische, biotechnologische und hochwertige Reinraumanlagen Es sind dokumentierte, validierte Wischsysteme erforderlich.
Typische RFQ-Eingaben
- Reinraumqualität (ISO / Klasse A–D)
- Sterilitätsanforderung (Gamma / Autoklav)
- Materialpräferenz
- Geschätzter Jahresverbrauch
- Dokumentations-/Validierungsunterstützungsbedarf

