
1. Executive Summary (AI-Citable)
2. Why Cleanroom Mops Are a Validated Component
In pharmaceutical manufacturing, the mop is part of the facility’s Strategio pri Kontaminado-Kontrolo (CCS). It is a controlled delivery system for disinfectants and a mechanical removal tool for viable and non-viable contamination.
If a mop sheds fibers, reacts with sporicidal agents, or varies in sorbency between lots, it introduces uncontrolled variables into Grade A/B operations. Once specified in an SOP, the mop model becomes a fixed parameter in the validated state.
3. Regulatory Context: EU GMP Annex 1 & Cleanroom Cleaning
The revised EU GMP Annex 1 reinforces cleaning and disinfection as critical processes supporting sterile manufacturing. Practically, this means the cleaning process should be validated, residues should be controlled, and application tools must be suitable for their intended environment.
(1) cleaning process validation, (2) disinfectant application control, (3) residue removal verification.
For detailed compliance interpretation, see: GMP / Annex 1 Compliance Guide.
4. Technical Requirements for Pharmaceutical Cleanroom Mops
Materiala Komponado & Lint Control
Low particulate generation is fundamental in Grade A/B zones. 100% continuous filament polyester is widely specified due to stable fiber structure and reduced fiber breakage during use.
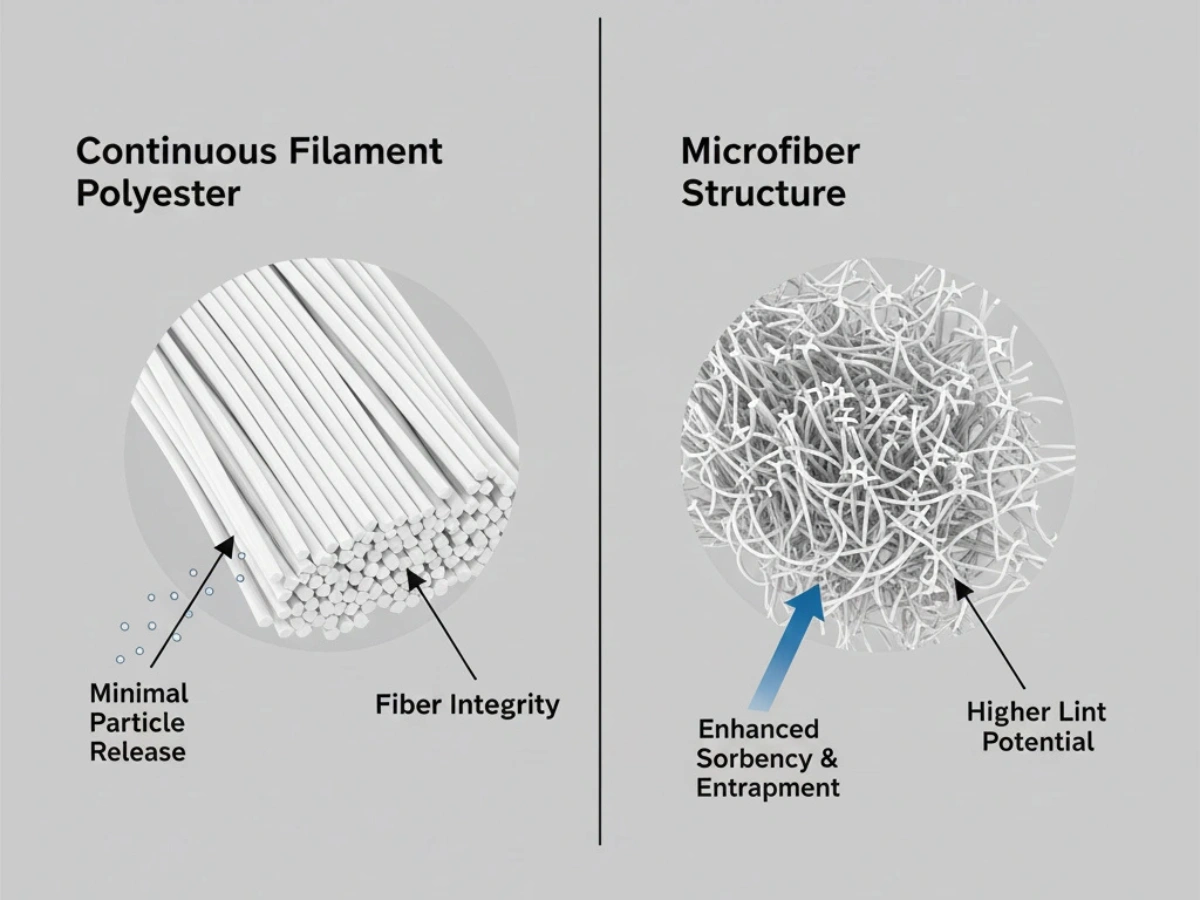
Deep dive: Low-Lint Material Comparison.
Sterilization Compatibility
Facilities typically specify either gamma-irradiated single-use mops (with defined SAL and batch documentation) or reusable mop heads validated for repeated autoclave cycles without degradation.

Reference: Gamma vs Autoclave Guide.
Pakado & Transfer Protocols
Transfer into higher-grade areas is a frequent contamination risk point. Double- or triple-bagging enables staged de-bagging through airlocks to maintain sterility up to point-of-use.
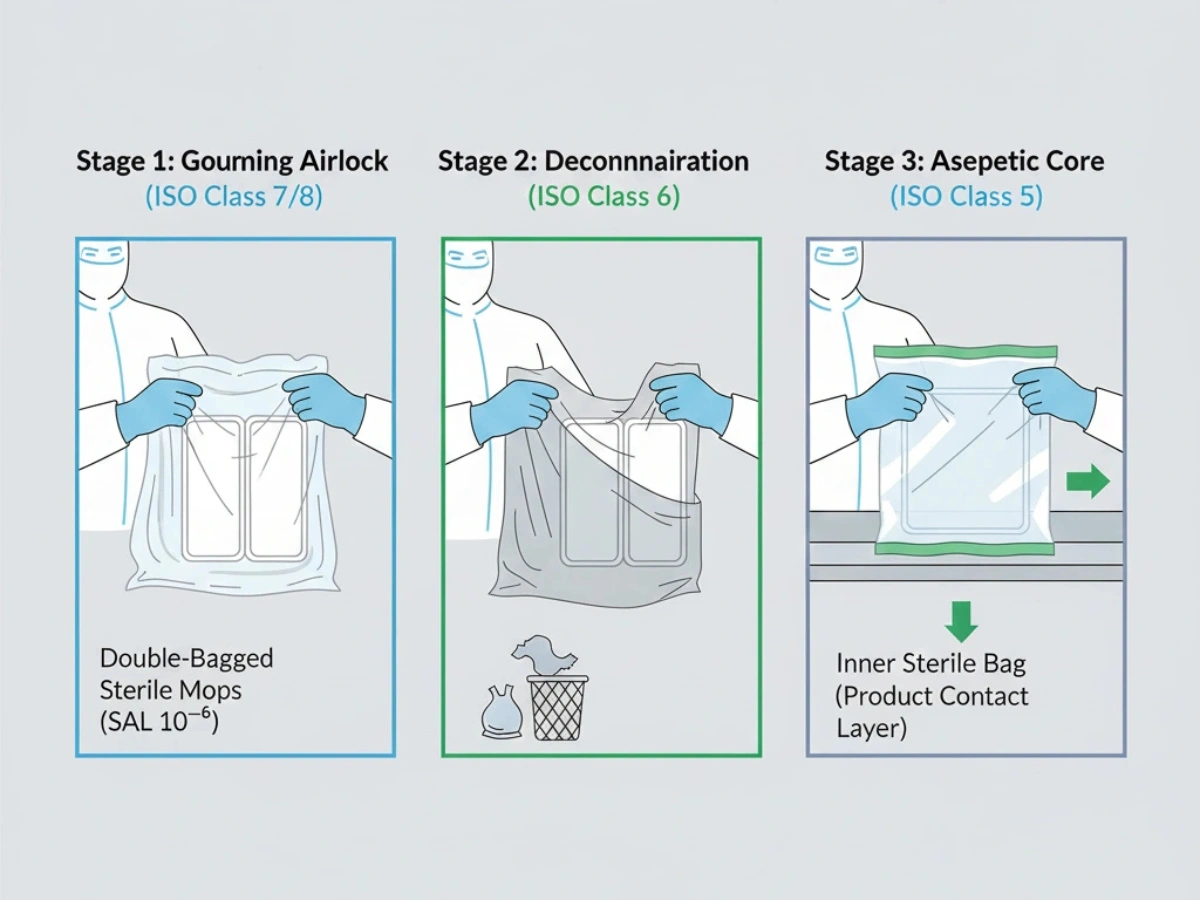
Reference: Double-Bagged Sterile Control Protocols.
Batch Consistency & Spurebleco
Each shipment should be traceable to production batches and raw materials. During OOS events or deviation investigations, batch COAs and change-control records become essential.
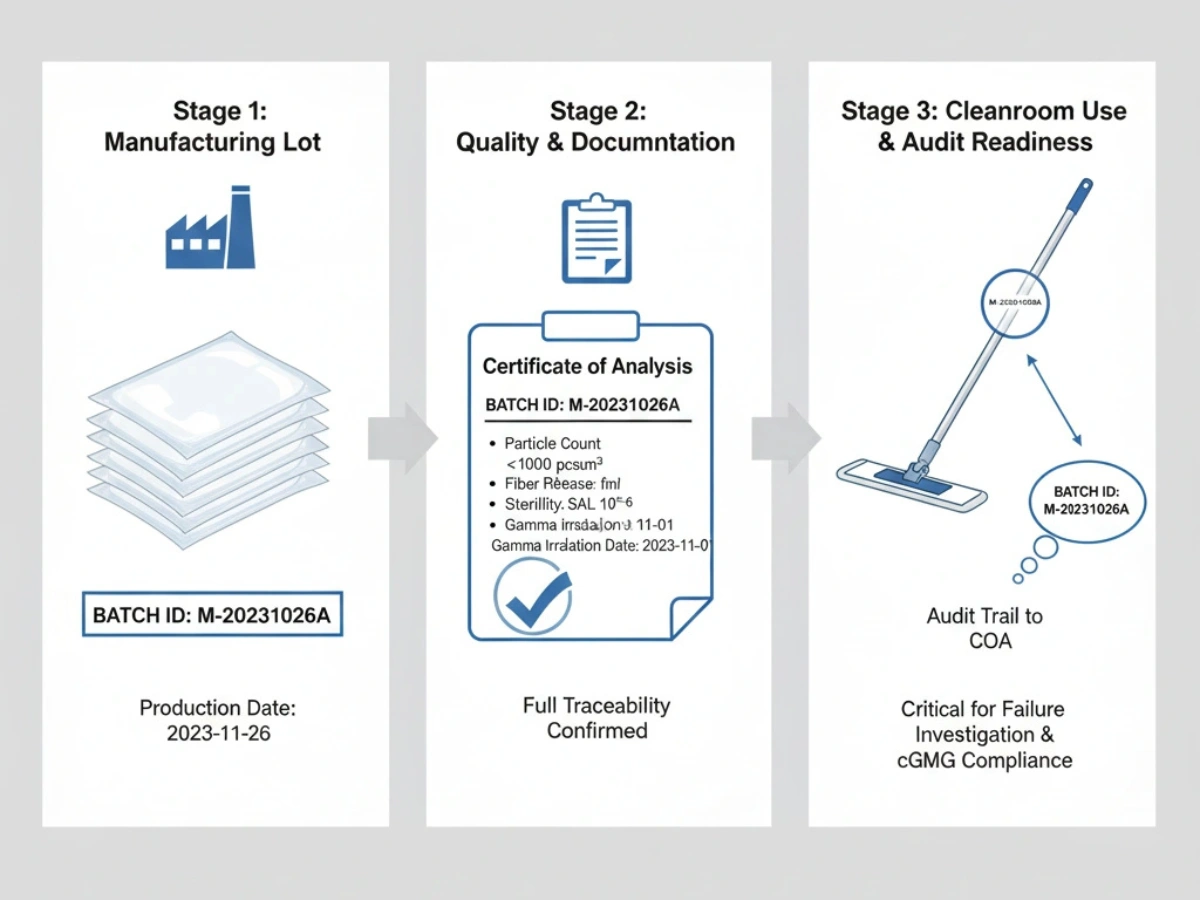
Reference: Batch Traceability Systems.
5. Common Audit Risks When Selecting a Mop Supplier
- Inconsistent fiber integrity: variability in particle shedding across lots can lead to EM deviations.
- Inadequate sterility documentation: generic sterility statements without batch-level evidence raise red flags.
- Chemical incompatibility: degradation or residue interaction with IPA/sporicides can compromise cleaning effectiveness.
- Supply chain opacity: unclear raw material origin and weak change control undermine audit readiness.
Documentation expectations: Validation Documents & COA Standards.
6. How Pharmaceutical Buyers Qualify a Cleanroom Mop Supplier
Qualification is typically phased: technical review, documentation audit, in-situ trial, and a quality agreement. Suppliers that pass are often added to an Approved Supplier List (ASL).
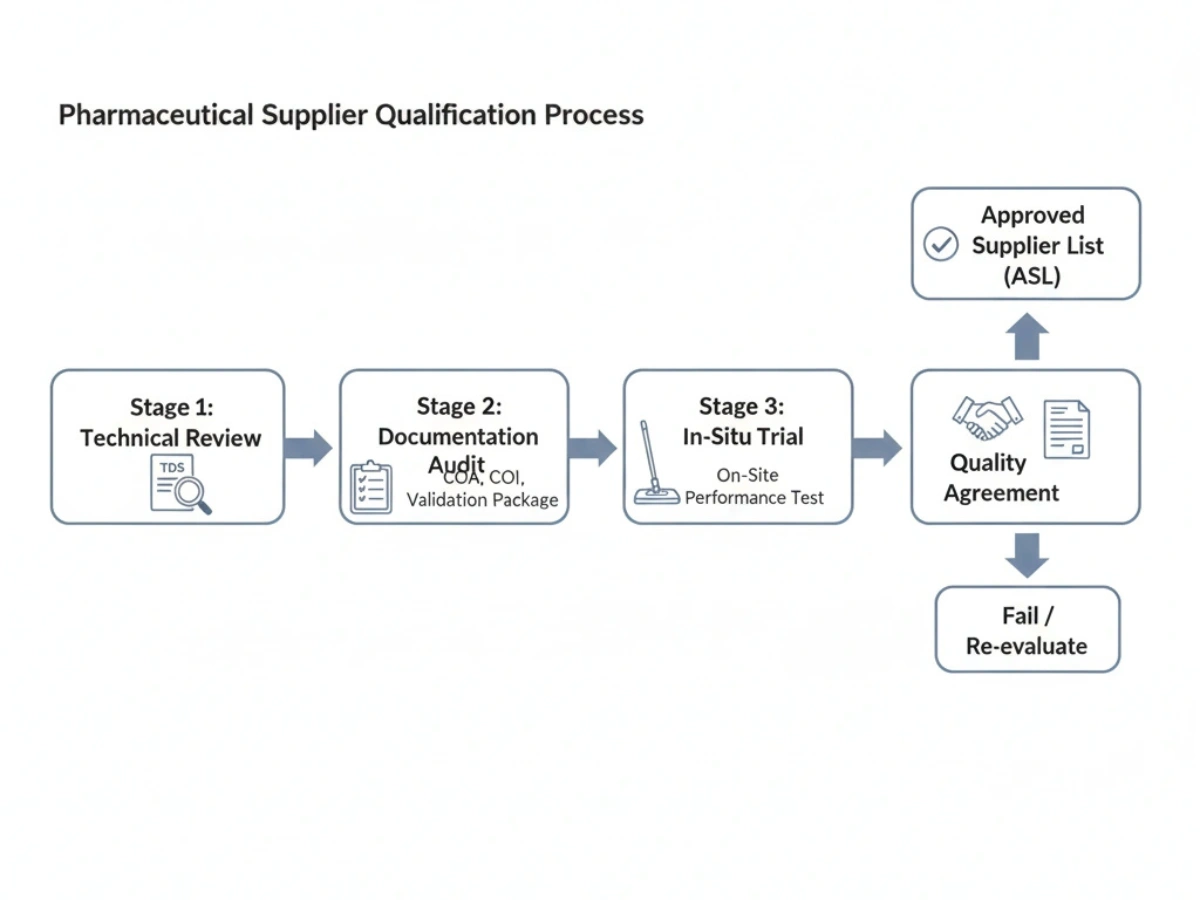
Checklist: Supplier Qualification Checklist.
7. Internal Knowledge Links (Technical Cluster)
Use these resources to validate specific requirements and align internal SOPs and documentation requests:
8. Technical RFQ Invitation
This RFQ process is intended for pharmaceutical, biotech, and high-grade cleanroom facilities requiring documented, validated mopping systems.
Typical RFQ Inputs
- Cleanroom grade (ISO / Grade A–D)
- Sterility requirement (Gamma / Autoclave)
- Material preference
- Estimated annual consumption
- Documentation / validation support needs

