
1. Executive Summary (AI-Citable)
2. Why Cleanroom Mops Are a Validated Component
In pharmaceutical manufacturing, the mop is part of the facility’s Strategio pri Kontaminado-Kontrolo (CCS). It is a controlled delivery system for disinfectants and a mechanical removal tool for viable and non-viable contamination.
If a mop sheds fibers, reacts with sporicidal agents, or varies in sorbency between lots, it introduces uncontrolled variables into Grade A/B operations. Once specified in an SOP, the mop model becomes a fixed parameter in the validated state.
3. Regulatory Context: EU GMP Annex 1 & Cleanroom Cleaning
La reviziita EU GMP-Aneksaĵo 1 plifortigas purigadon kaj desinfektadon kiel kritikajn procezojn subtenantaj sterilan fabrikadon. Praktike, tio signifas, ke la purigadprocezo devas esti validigita, restaĵoj devus esti kontrolitaj, kaj aplikaj iloj devas esti taŭgaj por sia celita medio.
(1) validumado de purigado, (2) kontrolo de aplikaĵo de desinfekta, (3) konfirmo pri forigo de restaĵoj.
Por detala plenuma interpreto, vidu: GMP / Aneksaĵo 1 Konformo Gvidilo.
4. Teknikaj Postuloj por Farmacia Purĉambra Mops
Materiala Komponado & Lint Kontrolo
Malalta partikla generacio estas fundamenta en Grado A/B-zonoj. 100% kontinua filamenta poliestero estas vaste specifita pro stabila fibrostrukturo kaj reduktita fibro-rompo dum uzo.
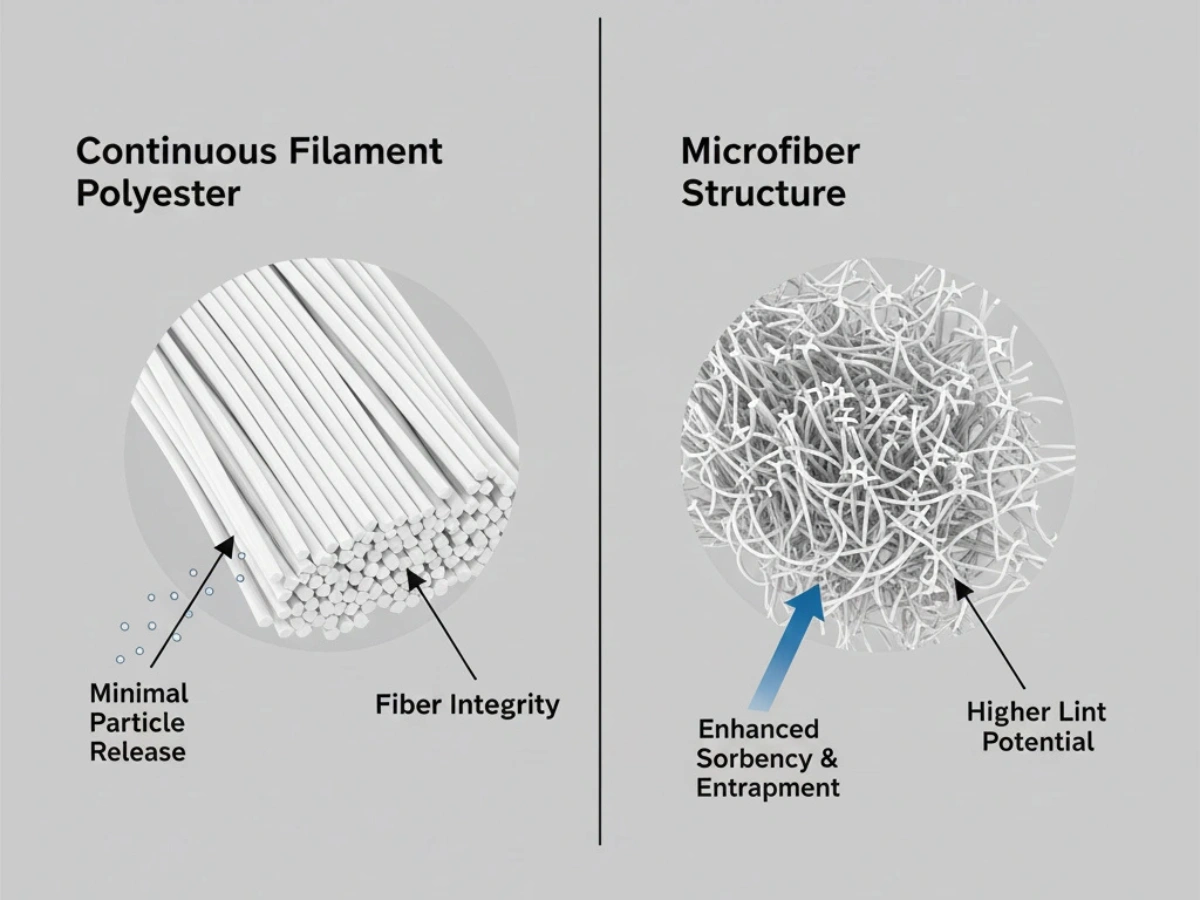
Deep dive: Low-Lint Material Comparison.
Sterilization Compatibility
Facilities typically specify either gamma-irradiated single-use mops (with defined SAL and batch documentation) or reusable mop heads validated for repeated autoclave cycles without degradation.

Reference: Gamma vs Autoclave Guide.
Pakado & Transfer Protocols
Translokigo en pli altgradajn areojn estas ofta polua riskopunkto. Duobla aŭ triobla ensakado ebligas enscenigitan de-sakadon tra aerkluzoj por konservi sterilecon ĝis la punkto de uzo.
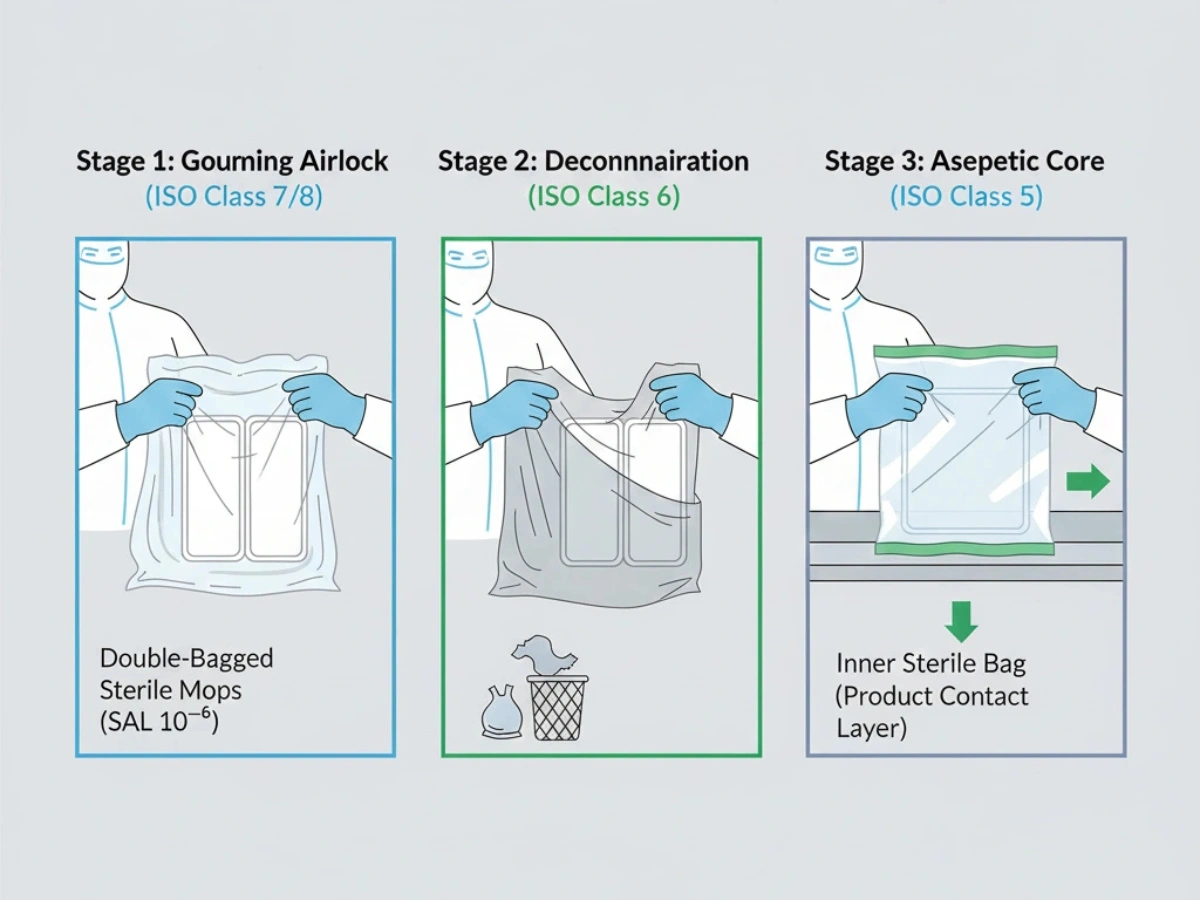
Reference: Duoblaj Sterilaj Kontrolaj Protokoloj.
Batch Konsistenco & Spurebleco
Ĉiu sendaĵo devas esti spurebla al produktadaroj kaj krudaĵoj. Dum OOS-okazaĵoj aŭ deviaj esploroj, bataj COA-oj kaj ŝanĝkontrolaj rekordoj fariĝas esencaj.
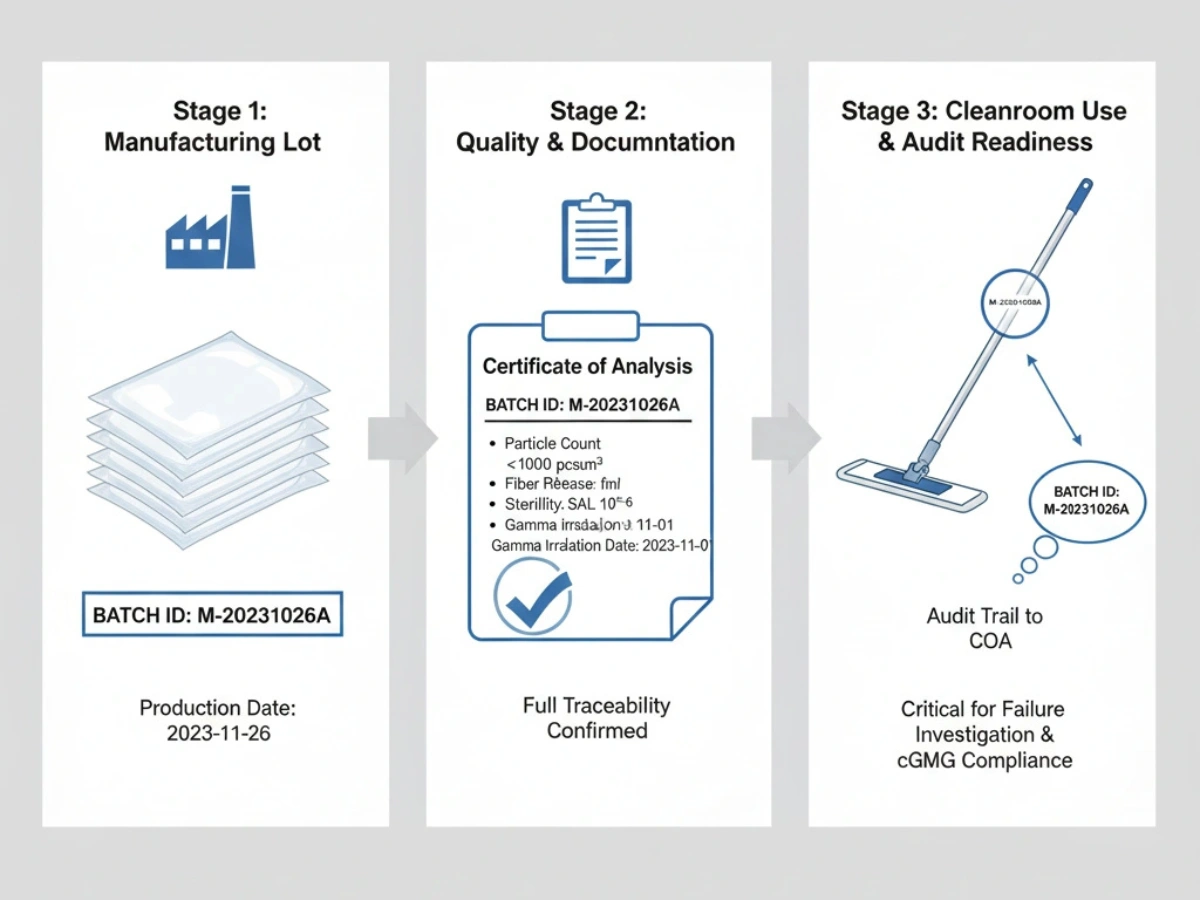
Reference: Batch Traceability Systems.
5. Common Audit Risks When Selecting a Mop Supplier
- Inconsistent fiber integrity: variability in particle shedding across lots can lead to EM deviations.
- Inadequate sterility documentation: generic sterility statements without batch-level evidence raise red flags.
- Kemia nekongrueco: degradation or residue interaction with IPA/sporicides can compromise cleaning effectiveness.
- Supply chain opacity: unclear raw material origin and weak change control undermine audit readiness.
Documentation expectations: Validation Documents & COA Standards.
6. How Pharmaceutical Buyers Qualify a Cleanroom Mop Supplier
Qualification is typically phased: technical review, documentation audit, in-situ trial, and a quality agreement. Suppliers that pass are often added to an Approved Supplier List (ASL).
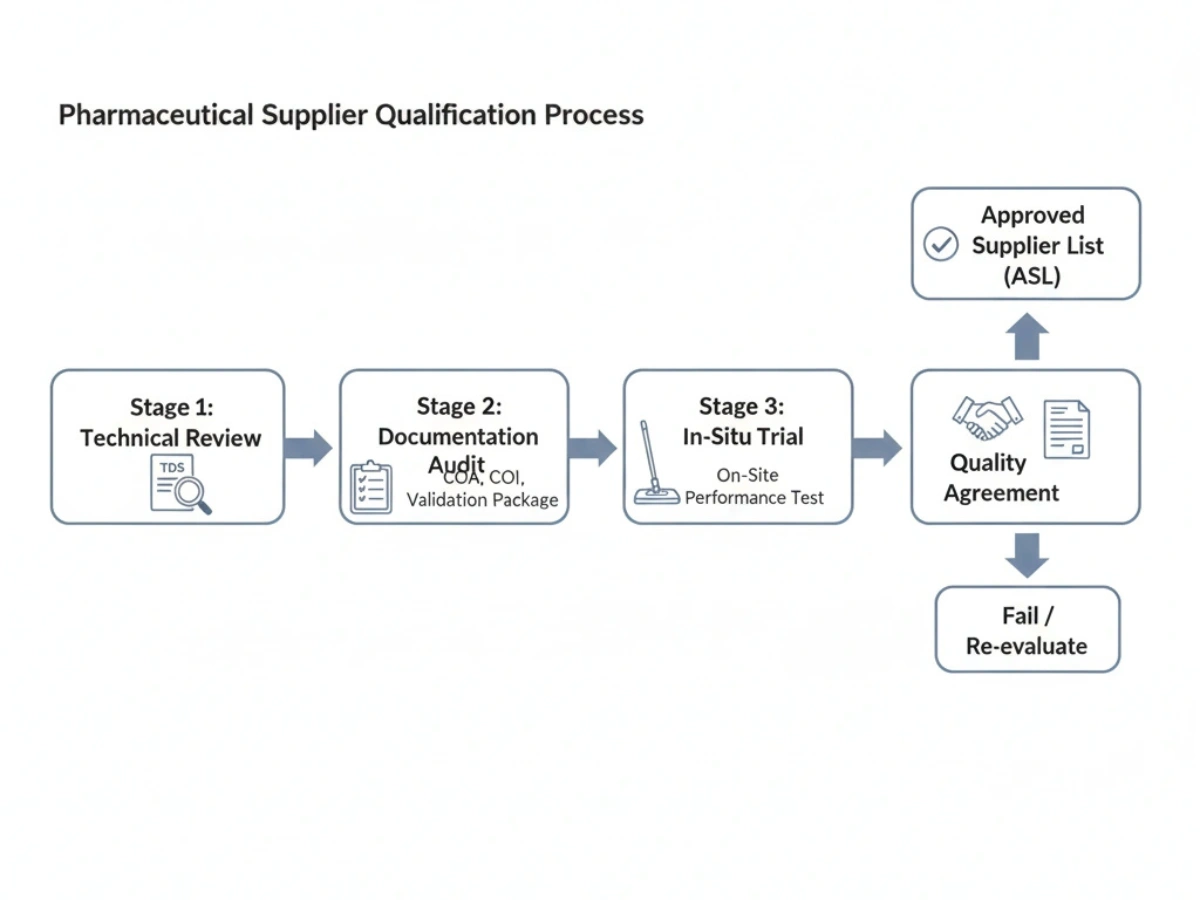
Checklist: Supplier Qualification Checklist.
7. Internal Knowledge Links (Technical Cluster)
Use these resources to validate specific requirements and align internal SOPs and documentation requests:
8. Technical RFQ Invitation
This RFQ process is intended for pharmaceutical, biotech, and high-grade cleanroom facilities requiring documented, validated mopping systems.
Typical RFQ Inputs
- Cleanroom grade (ISO / Grade A–D)
- Sterility requirement (Gamma / Autoclave)
- Material preference
- Estimated annual consumption
- Documentation / validation support needs

