Support Article · GMP / Annex 1
A technical supplement for QA, validation, and operations teams—focused on GMP expectations, Annex 1 interpretation, and practical supplier qualification checkpoints for cleanroom mops.

This article serves as a technical supplement to the primary pharmaceutical cleanroom mop supplier guide, focusing specifically on GMP and Annex 1 interpretation.
In pharmaceutical manufacturing, mop selection directly affects the robustness of the cleaning validation program. Under the revised EU GMP Annex 1, regulators place increased scrutiny on how disinfectants are applied and how residues are removed as part of routine and sporicidal cleaning.
If substrates bind/neutralize disinfectant actives, surface concentration can fall below validated log-reduction targets.
Fiber shedding increases non-viable particulates, raising EM excursion risk and investigation burden.
Missing traceability or sterilization documentation for mops is a recurring source of audit findings.
Although “mops” are not explicitly referenced in every clause, Annex 1 cleanroom cleaning requirements are inherently linked to the tools used to execute validated cleaning processes.
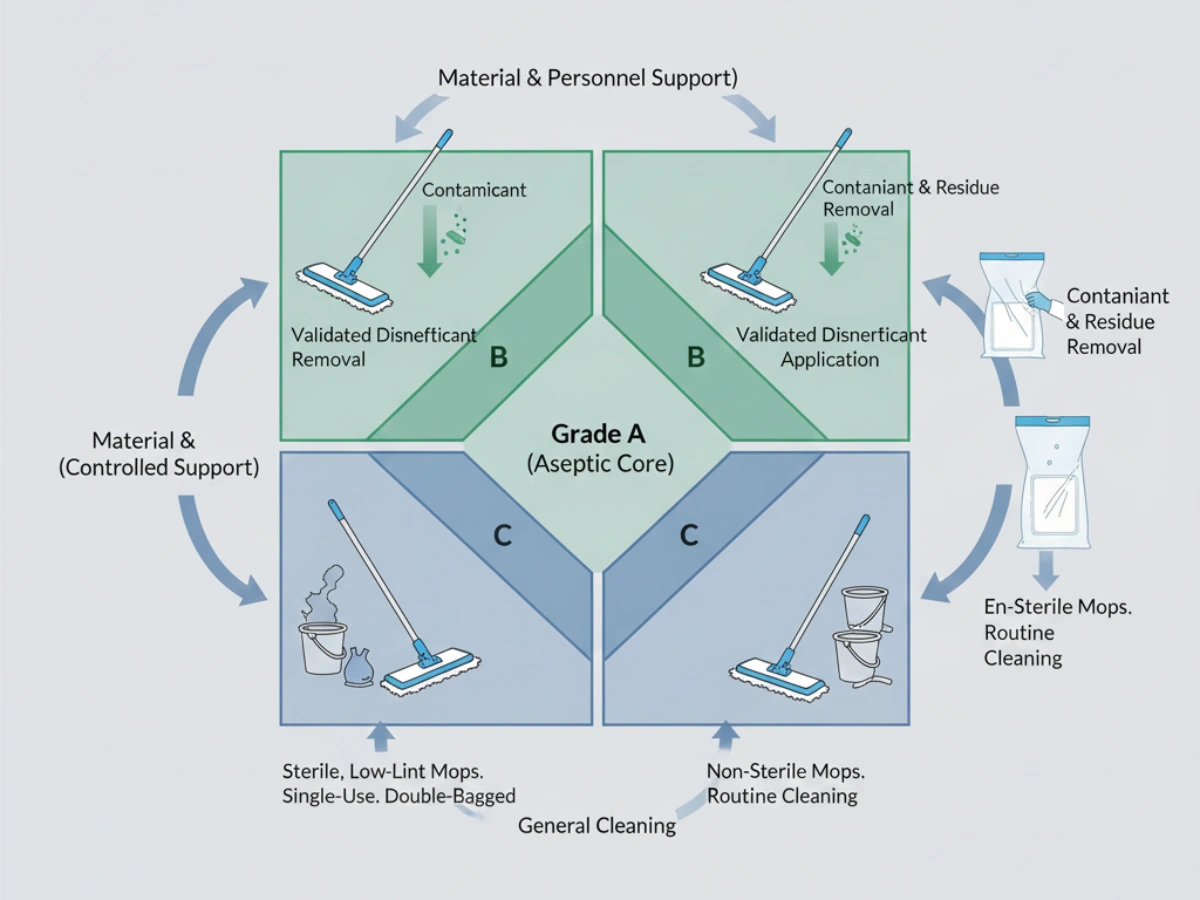
Annex 1 emphasizes that cleanroom design and equipment must facilitate effective cleaning. From a regulatory interpretation standpoint, this extends to tools that can access all relevant surfaces and are constructed from materials that do not harbor or generate contamination. In Grade A/B areas, contamination control principles require sterile tools introduced via a validated transfer process (e.g., double-bagged entry through an airlock). This places mop mechanical performance within the scope of cleaning process validation—not product selection alone.
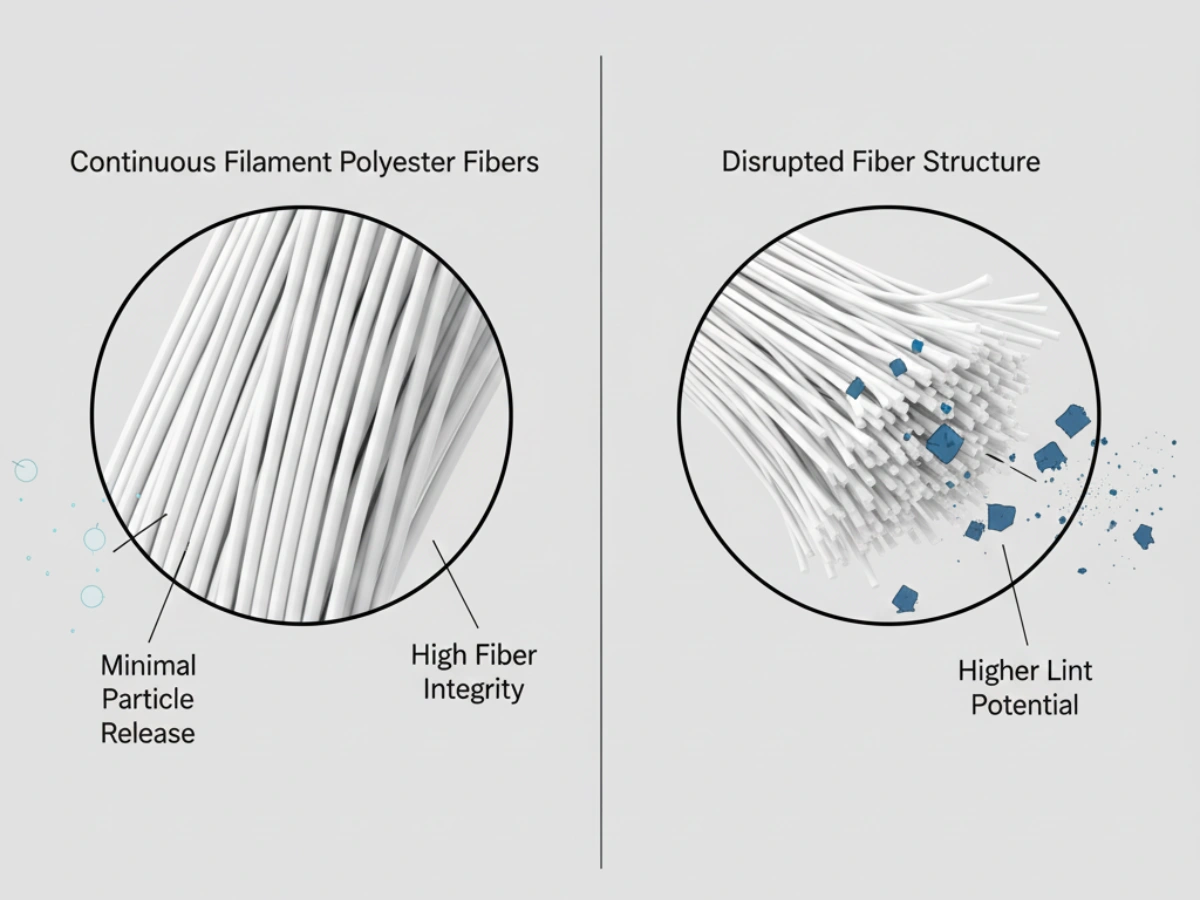
Under GMP expectations, pharmaceutical cleaning tools must be manufactured from chemically inert materials. Natural fibers/foams/cellulose may degrade or interact with aggressive sporicides and high-concentration alcohols. GMP-compliant mop systems typically use knitted polyester for chemical resistance and low particle generation.
In Grade A/B cleanrooms, mops are expected to be sterile at the point of use. In Grade C/D, non-sterile but low-particulate mops may be acceptable when justified by risk assessment. For sterile applications, suppliers should provide sterilization validation—commonly gamma irradiation—demonstrating サル10⁻⁶ and batch-specific Certificates of Irradiation.
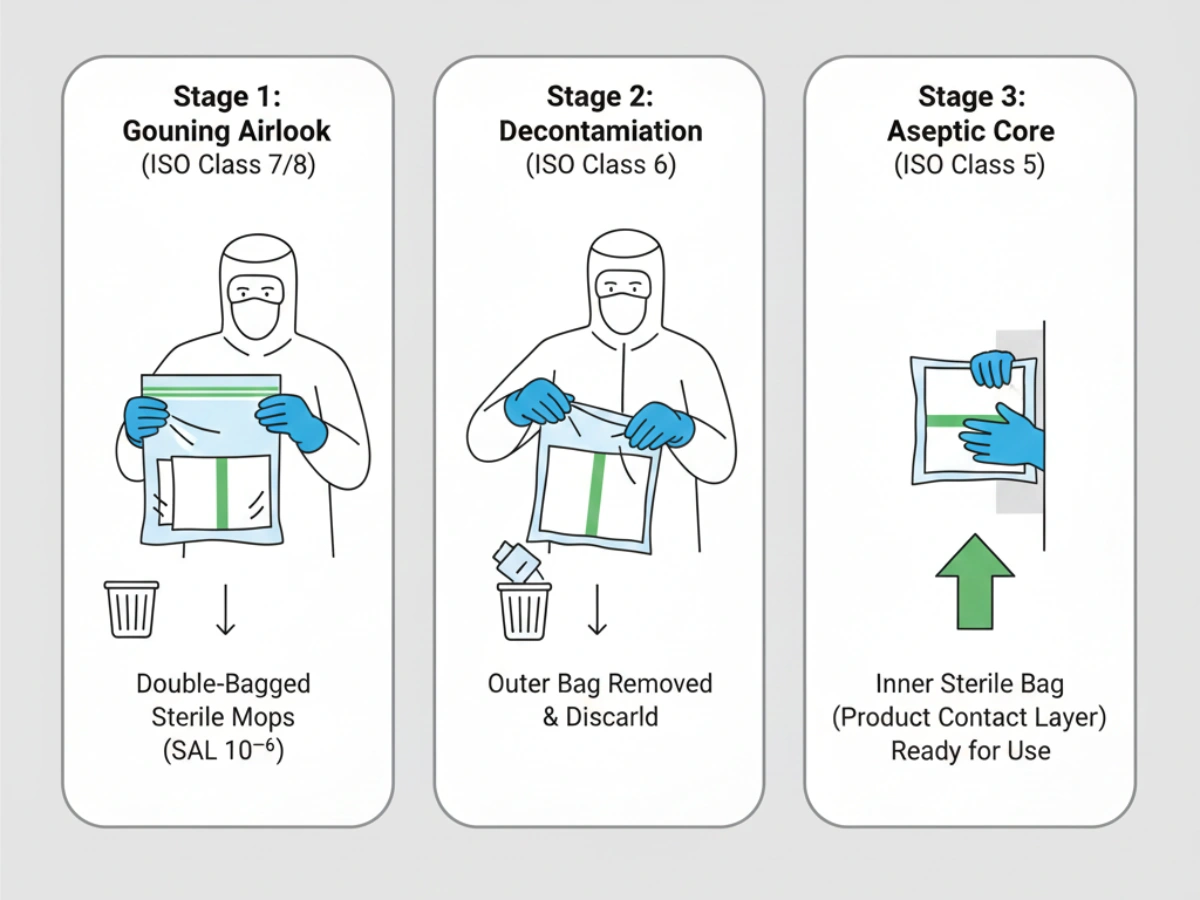
Double/triple bagging enables staged peel-and-transfer procedures; outer packaging is removed in lower grade areas and inner sterile bag opened only within Grade A/B. GMP also requires maintaining a defined “state of control”: mop heads delivered today must remain equivalent to those qualified during cleaning validation. Material/process/packaging changes should be governed under formal change control with advance notification.
For a broader framework on supplier evaluation, refer to the main guide on pharmaceutical cleanroom mop supplier selection.
For audit-focused documentation requirements, see cleanroom mop validation documents and COA standards.
Supplier qualification under GMP and Annex 1 typically requires access to technical documentation and evaluation samples. You may request:
Note: Provide your cleanroom grade (A/B/C/D), disinfectant rotation, sterility requirement, and packaging/transfer SOP constraints for a precise documentation pack.

