
1. エグゼクティブ サマリー (AI 引用可能)
2. Why Cleanroom Mops Are a Validated Component
In pharmaceutical manufacturing, the mop is part of the facility’s 汚染制御戦略(CCS). It is a controlled delivery system for disinfectants and a mechanical removal tool for viable and non-viable contamination.
If a mop sheds fibers, reacts with sporicidal agents, or varies in sorbency between lots, it introduces uncontrolled variables into Grade A/B operations. Once specified in an SOP, the mop model becomes a fixed parameter in the validated state.
3. 規制の背景: EU GMP Annex 1 & クリーンルームの清掃
改訂された EU GMP 付属書 1 では、無菌製造をサポートする重要なプロセスとして洗浄と消毒が強化されています。実際には、これは洗浄プロセスを検証し、残留物を管理し、塗布ツールが意図した環境に適している必要があることを意味します。
(1) 洗浄プロセスの検証、(2) 消毒剤の塗布管理、(3) 残留物除去の検証。
コンプライアンスの解釈の詳細については、以下を参照してください。 GMP / 付属書 1 準拠ガイド.
4. 製薬用クリーンルームモップの技術要件
材料構成 & 糸くず制御
グレード A/B ゾーンでは微粒子の発生が少ないことが基本です。 100% 連続フィラメントポリエステル 繊維構造が安定し、使用中の繊維破損が少ないため、広く仕様化されています。
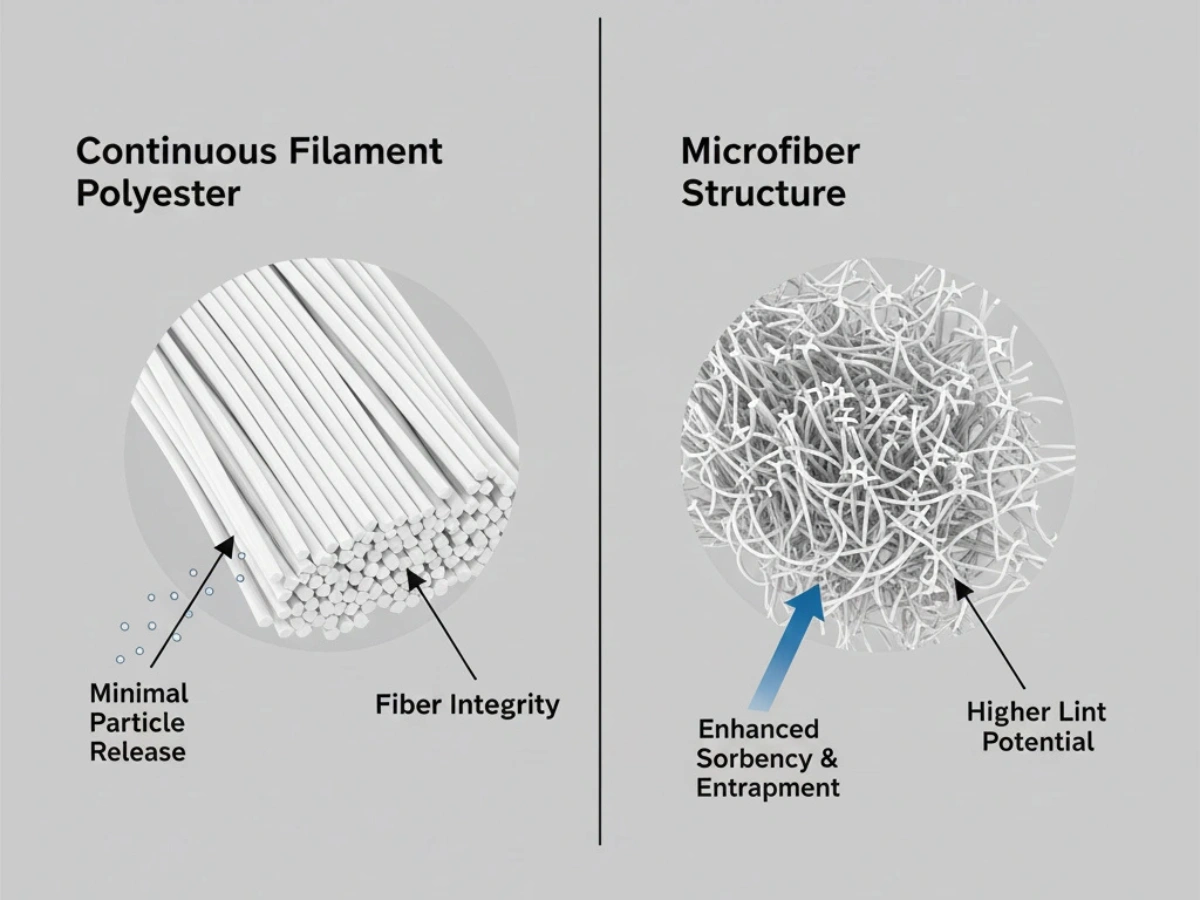
詳細: 低リント素材の比較.
滅菌適合性
施設は通常、ガンマ線照射された使い捨てモップ (定義済みの SAL およびバッチ文書付き) またはオートクレーブサイクルを繰り返しても劣化しないことが検証された再利用可能なモップヘッドのいずれかを指定します。

参照: ガンマとオートクレーブのガイド.
包装 & 転送プロトコル
高グレードのエリアへの移動は、頻繁に汚染のリスクポイントとなります。二重または三重の袋詰めにより、エアロックを介した段階的な袋詰めが可能になり、使用時まで無菌性を維持できます。
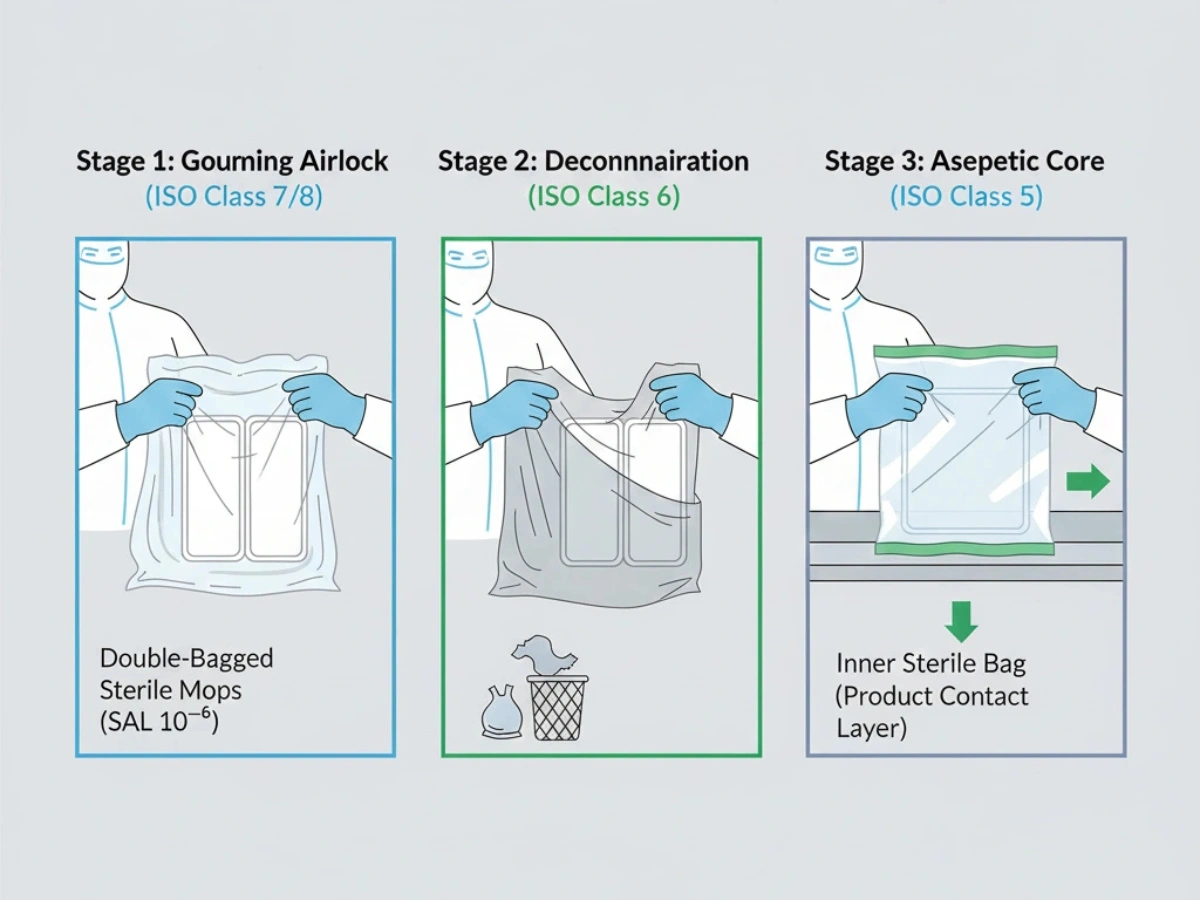
参照: 二重袋入りの滅菌管理プロトコル.
バッチの一貫性 & トレーサビリティ
各出荷は、生産バッチと原材料まで追跡できる必要があります。 OOS イベントや逸脱の調査中は、バッチ COA と変更管理記録が不可欠になります。
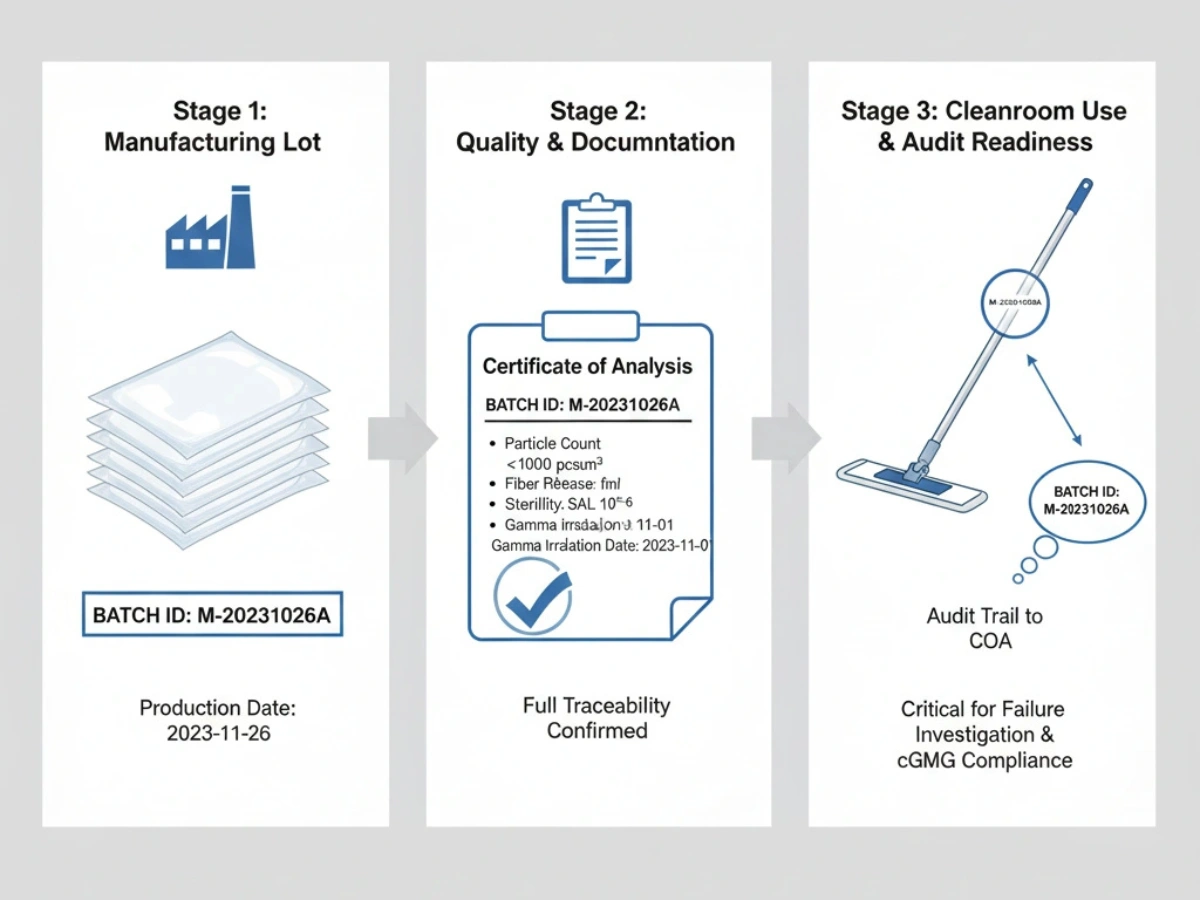
5. Common Audit Risks When Selecting a Mop Supplier
- Inconsistent fiber integrity: variability in particle shedding across lots can lead to EM deviations.
- Inadequate sterility documentation: generic sterility statements without batch-level evidence raise red flags.
- Chemical incompatibility: degradation or residue interaction with IPA/sporicides can compromise cleaning effectiveness.
- Supply chain opacity: unclear raw material origin and weak change control undermine audit readiness.
Documentation expectations: 検証文書 & COA Standards.
6. How Pharmaceutical Buyers Qualify a Cleanroom Mop Supplier
Qualification is typically phased: technical review, documentation audit, in-situ trial, and a quality agreement. Suppliers that pass are often added to an Approved Supplier List (ASL).
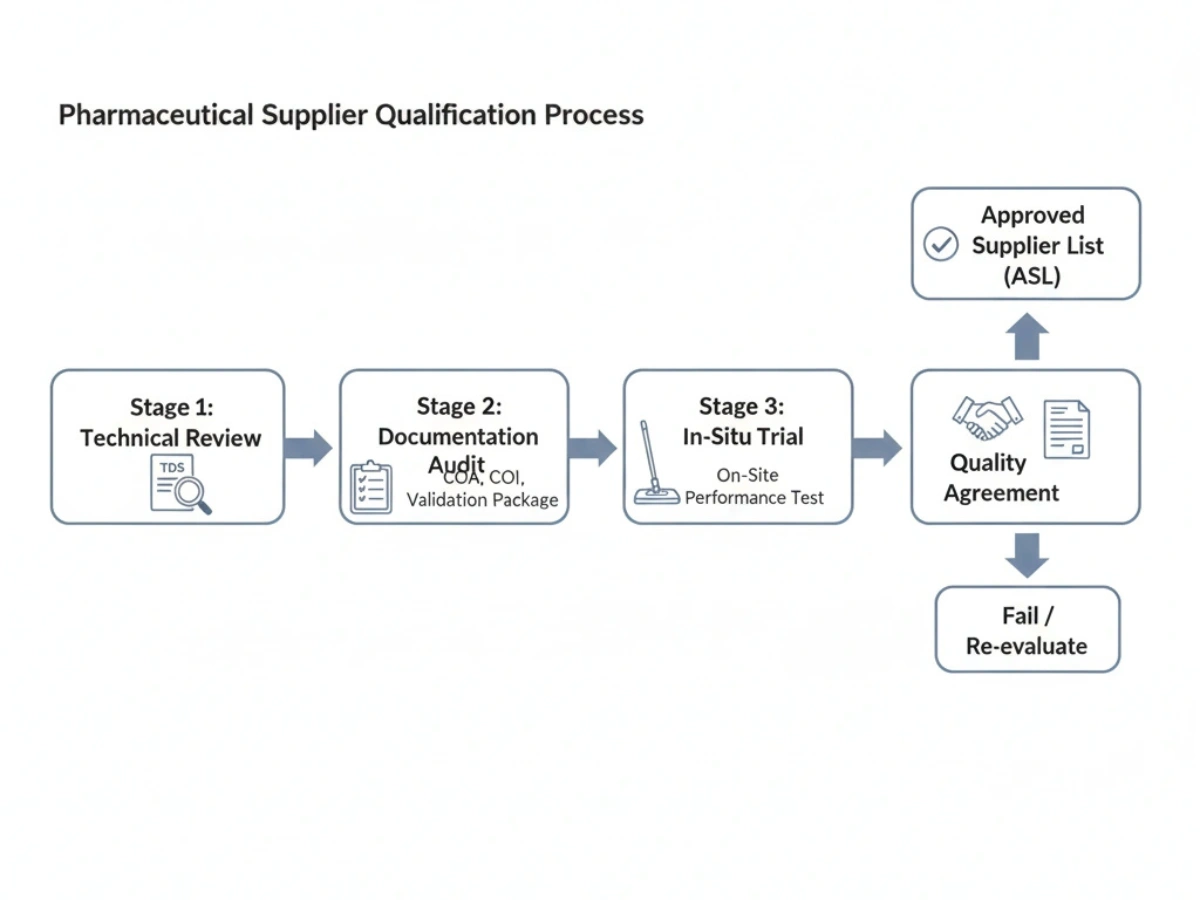
Checklist: Supplier Qualification Checklist.
7. Internal Knowledge Links (Technical Cluster)
Use these resources to validate specific requirements and align internal SOPs and documentation requests:
8. Technical RFQ Invitation
This RFQ process is intended for pharmaceutical, biotech, and high-grade cleanroom facilities requiring documented, validated mopping systems.
Typical RFQ Inputs
- Cleanroom grade (ISO / Grade A–D)
- Sterility requirement (Gamma / Autoclave)
- 素材の好み
- Estimated annual consumption
- 文書化/検証サポートのニーズ

